

| Gastroenterology Research, ISSN 1918-2805 print, 1918-2813 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Gastroenterol Res and Elmer Press Inc |
| Journal website http://www.gastrores.org |
Review
Volume 12, Number 5, October 2019, pages 221-232
Diagnosis and Treatment of Genetic HFE-Hemochromatosis: The Danish Aspect
Nils Thorm Milmana, d, Frank Vinholt Schioedta, Anders Ellekaer Junkerb, Karin Magnussenc
aDigestive Disease Center K, Bispebjerg Hospital, University of Copenhagen, Copenhagen, Denmark
bGastrounit, Medical Division, Hvidovre Hospital, University of Copenhagen, Copenhagen, Denmark
cDepartment of Blood Center and Medical Biochemistry, Innlandet Hospital Trust, Lillehammer, Norway
dCorresponding Author: Nils Thorm Milman, Lindevangen 87B, Virum DK-2830, Denmark
Manuscript submitted July 30, 2019, accepted August 28, 2019
Short title: HFE-Hemochromatosis in Denmark
doi: https://doi.org/10.14740/gr1206
- Abstract
- Introduction
- HFE-Associated Hemochromatosis
- Prevalence
- Pathogenesis
- Estimation of Body Iron Status
- Penetrance
- Symptoms
- Organ Involvement
- Diagnostic Evaluation and Examinations for HFE-Hemochromatosis
- Paraclinical Examinations
- Treatment
- Prognosis
- Diet and Dietary Supplements
- Family Screening
- Population Screening
- Conclusions
- References
| Abstract | ▴Top |
This paper outlines the Danish aspects of HFE-hemochromatosis, which is the most frequent genetic predisposition to iron overload in the five million ethnic Danes; more than 20,000 people are homozygous for the C282Y mutation and more than 500,000 people are compound heterozygous or heterozygous for the HFE-mutations. The disorder has a long preclinical stage with gradually increasing body iron overload and eventually 30% of men will develop clinically overt disease, presenting with symptoms of fatigue, arthralgias, reduced libido, erectile dysfunction, cardiac disease and diabetes. Subsequently the disease may progress into irreversible arthritis, liver cirrhosis, cardiomyopathy, pancreatic fibrosis and osteoporosis. The effective standard treatment is repeated phlebotomies, which in the preclinical and early clinical stages ensures a normal survival rate. Early detection of the genetic predisposition to the disorder is therefore important to reduce the overall burden of clinical disease. Population screening seems to be cost-effective and should be considered.
Keywords: Arthritis; Diabetes mellitus; Hemochromatosis type 1; HFE-associated hemochromatosis; Hereditary hemochromatosis; Hepatocellular carcinoma; Iron overload; Liver cirrhosis
| Introduction | ▴Top |
Genetic hemochromatosis is the term for mutations in various genes, all of which produce an increased intestinal iron uptake, which in the long term may cause accumulation of iron in the body organs. Genetic hemochromatosis is divided into two main groups: HFE-hemochromatosis and non-HFE-hemochromatosis. Among people of Northwestern European descent including ethnic Danes, HFE-hemochromatosis is by far the most common, while non-HFE hemochromatosis occurs sporadically [1].
The Danish National Board of Health in 2017 assigned the handling (evaluation, diagnosis and treatment) of patients with hemochromatosis to the specialty of gastroenterology and hepatology thereby terminating many years of frustration in these “homeless” patients, who, due to their plethora of symptoms, are referred from one specialty to another in order to obtain a diagnosis. This review is based on the Danish National Guidelines for HFE-hemochromatosis elaborated by the Danish Society for Gastroenterology and Hepatology [2]. The figures and text boxes are reproduced with permission from the Danish medical journal Ugeskrift for Laeger [3].
| HFE-Associated Hemochromatosis | ▴Top |
HFE-associated hemochromatosis is due to mutations, or variants, on the HFE-gene located on chromosome 6p. Most patients with clinical hemochromatosis are homozygous for the Cys282Tyr (C282Y) mutation, but other mutations such as His63Asp (H63D) and Ser65Cys (S65C) occur in many people. In ethnic Danes, the allele frequency of C282Y is 5.6%, of H63D 12.8% and of S65C 1.8%, respectively. The frequency of heterozygosity is the allele frequency multiplied by two (i.e. 12.2% for C282Y, 25.6% for H63D and 3.6% for S65C). The mutations and the predisposition to disease are inherited in an autosomal recessive way [4].
| Prevalence | ▴Top |
HFE-hemochromatosis is defined by the presence of homozygosity or compound heterozygosity for one or more of the known HFE-mutations. HFE-hemochromatosis is the most common genetic predisposition to iron overload disease among ethnic Danes and in people of Northern European descent [5]. In addition, homozygosity for the C282Y mutation is the leading cause of clinical hemochromatosis in ethnic Danes, that is more than 95% of patients with clinical hemochromatosis have this mutation [6]. Population studies have shown that among ethnic Danes, 0.4%, or 1 in 250, are C282Y homozygous equivalent to approximately 20,000 people in Denmark. In addition, more than 13% are heterozygous for C282Y and/or compound heterozygous for C282Y/H63D (1.4%), C282Y/S65C (0.1%) or H63D/S65C (0.4%) [7]. Based on these figures, the genetic prevalence of HFE-hemochromatosis among ethnic Danes is probably even higher than 0.4% and more than 500,000 people are compound heterozygous or heterozygous for the HFE-mutations [7]. In comparison, the UK Biobank Study examining more than 450,000 individuals of European descent found a prevalence of C282Y homozygosity of 0.6%, or 1 in 156 and a prevalence of C282Y heterozygocity of 14.3% [8].
| Pathogenesis | ▴Top |
High concentrations of iron in the cells and tissues will through the Fenton reaction trigger the formation of free hydroxyl radicals, which are toxic to the cells and can cause DNA damage and cell death with subsequent fibrosis and tissue remodeling [9, 10]. The pathogenetic mechanisms by which the mutated HFE-gene product afflicts the iron homeostasis are not fully understood. Hepcidin, which is produced in the liver, is the “master regulator” of body iron homeostasis, and its main task is to inactivate ferroportin [1]. Ferroportin has an important position in the regulation of iron transport out through the cell membrane (efflux) in enterocytes, hepatocytes and macrophages. Normal HFE- and transferrin receptor-2 complexes on the cell membrane of the hepatocytes stimulate the production/activation of hepcidin, which subsequently inhibits intestinal iron uptake. Conversely, iron deficiency leads to a reduced production of hepcidin, which causes an increased iron uptake [11]. In hemochromatosis, because of a defective HFE-complex, the production/activation of hepcidin is reduced, resulting in an increased intestinal iron uptake, which is largely independent of the body’s iron status. HFE-hemochromatosis is thus characterized by a low plasma concentration of hepcidin termed “hepcidin insufficiency” [1, 11].
The increased iron absorption results in a high serum or plasma iron and a high serum or plasma transferrin saturation. At transferrin saturation values > 60%, non-transferrin-bound iron occurs in plasma, which is readily taken up by the cells in various organs (e.g. liver, pancreas, heart and endocrine organs). However, one exception is the macrophages, which typically contain less iron than macrophages in normal control persons [11].
Intracellular iron accumulation triggers oxidative stress, DNA damage, cellular necrosis and over time fibrosis. This development is typically seen in the liver, where initial fibrosis may eventually progress into cirrhosis [9, 10].
| Estimation of Body Iron Status | ▴Top |
The serum or plasma ferritin concentration is presently the best biomarker we have for the body’s iron content, with 1 µg/L corresponding to body iron reserves of 7 - 7.5 mg, that is a ferritin of 1,000 µg/L corresponds to an iron excess of 7.0 - 7.5 g [1, 12, 13].
The serum transferrin saturation percentage is an indicator of the iron content of the blood and the iron supply to the organs and helps classify the type of genetic hemochromatosis [11, 14]. A high serum transferrin saturation is usually the first indicator of HFE-hemochromatosis and can be present, even though serum ferritin is still within the normal range. Ferritin gradually increases later in the course of the disease. A fasting transferrin saturation of ≥ 45% is defined as being elevated [15, 16] and should trigger a control measurement and an additional measurement of serum ferritin. If the saturation in two consecutive measurements is ≥ 45% and/or ferritin is elevated (≥ 200 µg/L in women, ≥ 300 µg/L in men), the person should be examined for the most common HFE-mutations C282Y, H63D and S65C.
| Penetrance | ▴Top |
The penetrance of the HFE-mutations depends on gender and age plus the interaction between the HFE-mutations and the composition of the other genes that control the body iron uptake [7]. In addition, external factors such as the iron content of the diet, the composition of the diet, that is the balance between promoters and inhibitors of iron absorption, alcohol consumption, intake of iron supplements and vitamin C (ascorbic acid) [14] as well as blood donations [12, 13] will affect the iron homeostasis and the penetrance [17]. The penetrance is moderate in premenopausal women, because approximately 50% of women of reproductive age in Denmark and other Northern European countries have low body iron reserves [18], mainly due to blood losses at menstruations and childbirths [19] but also due to a relatively low dietary iron intake when compared to the recommended intake [20]. However, after the menopause, the penetrance increases significantly in women. A distinction is made between a preclinical stage in which the person, despite a moderate iron overload, does not have symptoms, and a clinical stage with greater iron excess, which causes symptoms from various organs.
Biochemical penetrance
Biochemical penetrance is defined as an increased fasting transferrin saturation ≥ 45% and an elevated serum ferritin ≥ 200 µg/L in women and ≥ 300 µg/L in men. The biochemical penetrance among C282Y homozygous Danish men is high: 94% have serum ferritin ≥ 300 µg/L, and 44% have values > 800 µg/L. Among C282Y/H63D compound heterozygotes, penetrance is 1-2% [7]. Among H63D homozygotes, 16% have increased transferrin saturation and 29% have increased ferritin [21]. High biochemical penetrance is also seen in other populations of Northwestern European descent [22].
Clinical penetrance
Clinical penetrance is defined as biochemical penetrance plus symptoms and/or organ dysfunction. The clinical penetrance is high in men and lower in women. In a population of more than 31,000 40-69-year-old persons of Northern European descent, 203 were C282Y homozygous. Among these, 28.4% of men and 1.2% of women had iron overload disease [23]. Among C282Y/H63D compound heterozygotes, 1.2% of men (1 out of 82) had iron overload disease compared to none of women (0 out of 95) [22]. In general, C282Y/H63D compound heterozygotes are at low risk of hemochromatosis-related morbidity [8, 24]. Among H63D homozygotes, 6.7% have iron overload disease [21].
The UK Biobank Study showed that in a large community sample of European descent, during a lifetime, C282Y homozygosity is associated with substantial clinically diagnosed morbidity in both men and women [8]. C282Y homozygous men aged 40 - 70 years (n = 1,294) had a significantly higher prevalence of liver disease, rheumatoid arthritis, osteoarthritis, osteoporosis and diabetes mellitus, compared to men without C282Y mutations (n = 175,539) (P < 0.001). In women aged 40 - 70 years (n = 1,596) C282Y homozygosity was associated with osteoarthritis (P < 0.001) [8].
C282Y homozygous men (n = 1,312) aged 60 - 70 years had an increased likelihood of reporting chronic pain, frailty and diagnoses of polymyalgia rheumatica and sarcopenia, compared to the wild-type genotype. C282Y homozygote women (n = 312) aged 65 - 70 were more likely to be frail and have chronic knee, hip, and back pain. Overall, 1.5% of frail men and women in the 65-70-year age group were C282Y homozygous [25].
The overall penetrance (i.e. having a diagnosis of hemochromatosis, liver disease, rheumatoid arthritis, osteoarthritis or diabetes mellitus) was 38% in C282Y homozygous men, compared with 16% in men with no C282Y mutations (i.e. an excess morbidity of 22%). In women, the respective figures were 27% versus 17% (i.e. an excess morbidity of 10%) [8].
| Symptoms | ▴Top |
Persons with HFE-hemochromatosis are often discovered randomly in connection with routine blood sampling and approximately 75% are asymptomatic at the time of diagnosis [1, 7]. Most persons have no symptoms in the early stage of the disease. With increasing iron accumulation in various tissues and organs, the patients develop an increasing burden of symptoms and risk of organ involvement [26, 27] (Table 1). Thus, the clinical picture will vary depending on how early or how late the diagnosis is made and how early or how late iron-depletion treatment is started.
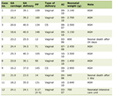 Click to view | Table 1. Patients’ Symptoms and Risk of Organ Involvement |
Symptoms are most often seen after the age of 30 years in men and after the menopause in women. Frequent onset symptoms are fatigue, arthralgias, abdominal pain as well as decreased libido and erectile dysfunction in men. In a Danish study of 179 patients, the first symptoms were fatigue (80%), arthralgias (65%) and decreased libido (40%) [27]. In a Norwegian study, the most common symptoms were also fatigue (46%), arthralgias (44%) and decreased libido (26%) [28]. Together, these symptoms have a negative impact on the quality of life. As fatigue is also a well-known symptom of iron deficiency, some patients start to take iron tablets either on their own initiative or at the recommendation of their doctor, without prior examination of their iron status [28, 29].
| Organ Involvement | ▴Top |
Liver
The excess iron predominantly accumulates in the hepatocytes. The first sign of liver dysfunction is persistently, mildly to moderately elevated liver enzymes alanine aminotransferase and aspartate aminotransferase. At ferritin concentrations above 700 - 800 µg/L, liver fibrosis begins to develop [26, 29]. If iron depletion treatment (phlebotomy) is not initiated, iron accumulation increases and the fibrosis may progress into liver cirrhosis, which subsequently may be complicated by hepatic insufficiency and hepatocellular carcinoma (HCC) [30]. Development of liver cirrhosis may also be triggered by other hepatic co-morbidities, such as alcoholic liver disease or viral hepatitis [30]. Cirrhosis is seen in patients with high ferritin concentrations above 1,000 µg/L. A ferritin level below 1,000 µg/L has a negative predictive value of 95% in the diagnosis of cirrhosis. Among randomly selected patients with liver cirrhosis, the prevalence of homozygosity for the C282Y mutation is 10-fold higher than in the background population, emphasizing the importance of screening all patients with liver cirrhosis for hemochromatosis [31].
The clinical and paraclinical pictures of liver cirrhosis associated with HFE-hemochromatosis do not differ from those of cirrhosis due to other causes. In a study of 120 cirrhosis patients with hemochromatosis versus 120 patients with cirrhosis due to other causes, there was a similar prevalence of esophageal varices and splenomegaly [32].
HCC is most often seen in patients with liver cirrhosis but may occur in some patients without cirrhosis [33, 34]. Among randomly selected patients with HCC, there is a high prevalence of C282Y homozygosity of 6-10%, emphasizing that all patients with HCC should be screened for hemochromatosis. The risk of developing HCC in cirrhosis due to HFE-hemochromatosis is 20-fold higher than that in the background population.
Extrahepatic cancer
The incidence of other cancers remains controversial. A Swedish study showed no increased incidence of extrahepatic cancer in a small series of relatives to patients with hemochromatosis [35]. Among Danish C282Y homozygous patients with transferrin saturation > 60%, an increased incidence of all cancers was found in both women and men, compared to the control group [36] and the authors of the study recommend that the clinician should give increased attention to the occurrence of cancer in patients with hemochromatosis [36]. An Australian cohort study involving 28,509 persons found an increased risk of colorectal cancer in women and men and an increased risk of breast cancer in women [37].
Heart
The myocardium is particularly sensitive to iron-induced oxidative stress due to the high content of mitochondria and a low content of antioxidants [38, 39]. As a consequence of iron-induced cardiomyopathy, restrictive diastolic dysfunction and various forms of arrhythmias are seen in the initial stage [39, 40]. Later, patients may develop ventricular arrythmias and overt heart failure, which previously was a frequent cause of death in patients with hemochromatosis [41].
Joints and bones
Arthritis occurs most frequently in the small finger joints, especially the second and third metacarpophalangeal joints and may display a characteristic radiological pattern [23], but larger joints can also be afflicted [27]. Typically, several joints are afflicted simultaneously [42, 43]. The pathogenesis remains unclear. Various grades of iron deposits are present in the synovial membrane but there is no straightforward association between the magnitude of iron overload and occurrence of arthritis. Arthritis has been described in several HFE-hemochromatosis patients without significant iron overload indicating that other factors than just iron per se may be involved [44] (e.g. yet unknown genetic factors associated with the C282Y mutation).
Osteopenia and osteoporosis occur frequently; among patients with clinical hemochromatosis 41% have osteopenia and 25% osteoporosis. The frequency of osteoporosis increases with increasing iron overload. The pathogenesis remains unclear; both increased bone resorption and reduced bone formation occur simultaneously [45].
Endocrine glands
Pancreas
Iron-induced destruction of the insulin-producing beta-cells, possibly in combination with insulin resistance due to liver damage, leads to the development of diabetes mellitus, which initially is non-insulin-dependent. As iron overload increases, the diabetes will subsequently become insulin-dependent [27, 37]. The frequency of diabetes depends on how early in the course of the disease the diagnosis is made and how soon iron depletion treatment is started. In previous patient series with clinical hemochromatosis [27, 43], the incidence of diabetes was high. The exocrine pancreatic function is not afflicted.
Pituitary gland
Iron can accumulate in all pituitary cells (gonadotropic, thyrotropic, somatotropic, lactotropic, corticotropic) and to various extents affect the function of these endocrine cells [46]. Panhypopituitarism, however, is rare [47]. The gonadotropic cells are the first to be afflicted, leading to secondary hypogonadism. Hypogonadism in premenopausal women is evidenced by decreased levels of luteotropic and follicle stimulating hormone, decreased libido and amenorrhea [47]. In men, decreased production of luteotropic and follicle stimulating hormone will result in a decreased production of testosterone as well as decreased spermatogenesis and cause reduced libido and erectile dysfunction. Furthermore, hypogonadism contributes to an increased risk of osteoporosis.
Thyroid gland
There is often considerable iron accumulation in the thyroid. Clinical and subclinical hypothyroidism occurs in patients with clinical hemochromatosis, but the prevalence is barely significantly increased compared to the background population [48].
Skin
In patients with advanced clinical hemochromatosis, the combination of iron deposition in the skin and concurrent stimulation of the melanin production by melanocytes can lead to excess skin pigmentation, most often in the form of a very sun tanned or greyish appearance, which in combination with diabetes mellitus previously was called “bronze diabetes” [27].
| Diagnostic Evaluation and Examinations for HFE-Hemochromatosis | ▴Top |
A diagnostic and therapeutic algorithm is shown in Figure 1. The initial examinations (general clinical examination and blood sample analyses) can be performed by all medical doctors including general practitioners, while more elaborate examinations should be performed in the specialist departments in clinics or hospitals.
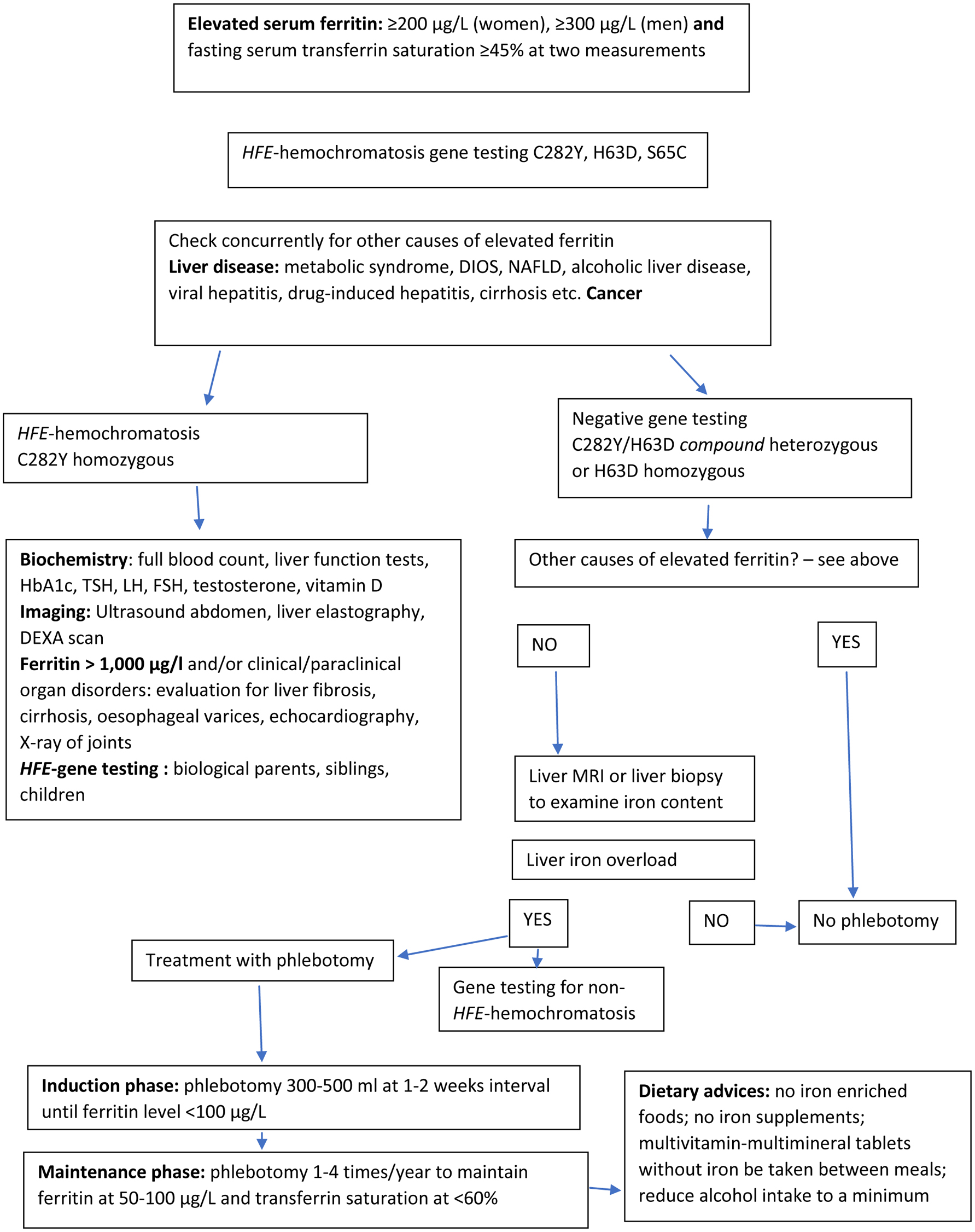 Click for large image | Figure 1. Diagnostic and therapeutic algorithm (simplified) in the assessment of HFE-hemochromatosis. |
| Paraclinical Examinations | ▴Top |
Liver biopsy with histochemical, semi-quantitative assessment or chemical determination of iron content was previously the standard diagnostic method in hemochromatosis. Following the introduction of the serum ferritin assays and HFE-genotyping, liver biopsy is now reserved for those patients where there is doubt about the diagnosis, such as in certain types of non-HFE hemochromatosis, dysmetabolic iron overload syndrome (DIOS), non-alcoholic fatty liver disease (NAFLD) [49], as well as some types of alcoholic liver disease presenting with elevated ferritin and moderate iron overload. However, patients with DIOS, NAFLD and alcoholic liver disease usually have a normal transferrin saturation, but an overlap with hemochromatosis is seen in some patients. In addition, liver biopsy may be used to assess the degree of fibrosis/cirrhosis and the presence of HCC.
Ultrasound scan of the liver is often the first diagnostic step,when the patient presents with elevated liver enzymes or there is suspicion of liver disease. Ultrasound cannot detect iron in liver tissue and therefore cannot be used for the diagnosis of iron overload in hemochromatosis [50, 51], but is useful for differential diagnostic purposes to exclude other causes of elevated liver enzymes and NAFLD. Ultrasound can also be used in the diagnosis of liver cirrhosis and HCC.
Ultrasound-based elastography (Fibroscan®) of the liver for assessment of fibrosis in hemochromatosis patients has only been validated in a few studies [51-53]. Further studies are needed to clarify whether this modality can be used in the investigation and follow-up of HFE-induced liver fibrosis.
Computed tomography (CT) of the liver can detect iron in the liver parenchyma, but the method requires special programming of the scanner, is semi-quantitative and has several sources of error [54, 55]. After the introduction of magnetic resonance imaging (MRI), CT scan is seldom used in the detection of liver iron content. However, focal lesions in the liver can be visualized by CT scan.
MRI scan of the liver is, in the experienced setting, a good method for quantitative measurement of the liver iron content [50, 56, 57]. There exists no common consensus on the methodology, so each center has to develop its own program and calibrate their scanner using objects with known iron concentrations. In Denmark, this modality is available at radiological centers in university hospitals.
MRI scan of the heart can be used to detect iron accumulation in the myocardium [41, 58] and is in Denmark performed at radiological centers in university hospitals. The iron content can also be assessed by histochemical methods in endomyocardial biopsies [59].
Electrocardiography and echocardiography are used in the screening for heart disease caused by HFE-hemochromatosis and other iron overload disorders [60]. Echocardiography and electrocardiography should always be performed when there are symptoms or suspicion of heart disease.
X-ray of the joints is used to evaluate the extent of arthritis. A rheumatological scoring system, based on X-rays of hands, wrists, knees and ankles, has been validated in a cohort of hemochromatosis patients with arthritis [61].
Dual-energy X-ray absorptiometry (DXA scan) is used to determine bone density and examine for osteopenia and osteoporosis [62].
| Treatment | ▴Top |
The standard treatment of HFE-hemochromatosis is simple, as the body’s excess iron is removed by repeated lettings of whole blood (phlebotomy) [63, 64]. It is an effective and low-cost procedure to reduce iron overload and there are few side effects. A patient-blinded, randomized study of patients with HFE-hemochromatosis and ferritin between 300 - 1,000 µg/L showed significant improvement in fatigue and cognitive scores in patients treated with erythrocyte-apheresis compared to patients treated with plasma-pheresis [65].
In normal persons, under conditions with a plentiful iron supply, the maximal erythropoietic response to anemia is approximately a 200% increase over basal erythropoietic rate [66]. In hemochromatosis patients, the erythropoiesis usually works in a normal way and most patients regain their habitual hemoglobin level in a few weeks. Prior to phlebotomy, the hemoglobin concentration should be checked. There is no consensus on which critical hemoglobin level should trigger postponement of phlebotomy. In Canada and USA, a critical value of 110 g/L (6.8 mmol/L) is recommended [17] and in Spain, a critical value of 120 g/L (7.4 mmol/L) is used (Milman et al, personal communication). When hemoglobin is below 120 g/L prior to phlebotomy, postponing the procedure should be considered until hemoglobin has increased. Subsequently extend the intervals between blood lettings and/or draw less than 450 - 500 mL blood at each procedure. The phlebotomy regime must be adjusted to the individual patient in order to avoid significant side effects, especially fatigue and lack of energy. Depending on the hemoglobin concentration, 220 - 300 mg of iron is removed by a bloodletting of 450 mL (Table 2).
Click to view | Table 2. Treatment of HFE-Hemochromatosis |
To the knowledge of the authors no studies have examined whether treatment with erythropoietin in order to stimulate erythropoiesis in patients with HFE-hemochromatosis may allow an increase in the phlebotomy rate in order to enhance the rate of body iron depletion.
Newly diagnosed patients start with “induction treatment”, consisting of phlebotomy at 1 - 2 week interval until ferritin has dropped to a level between 50 - 100 µg/L (Fig. 2).
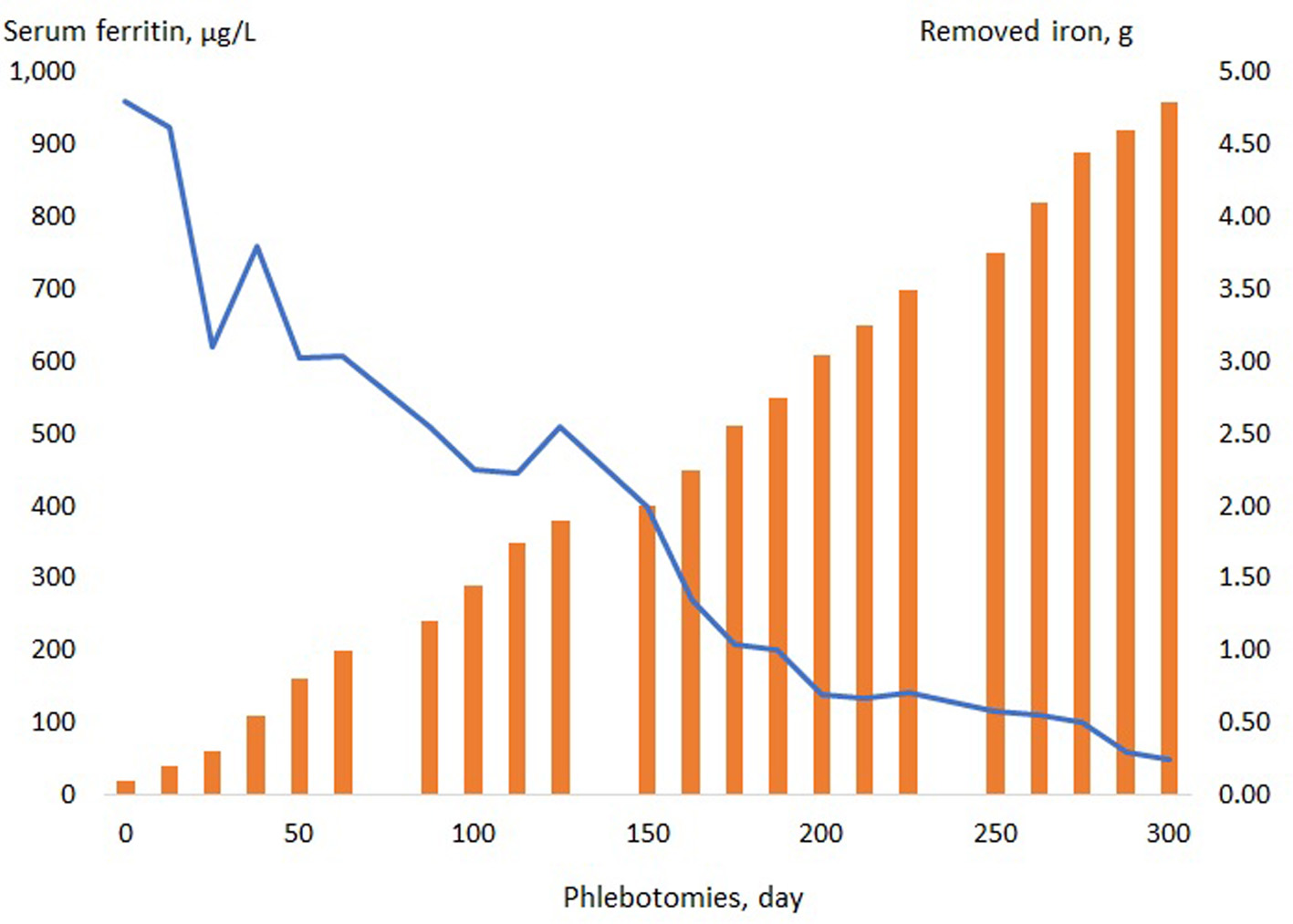 Click for large image | Figure 2. A 55-year-old woman with hemochromatosis and liver fibrosis. Due to fatigue, the patient had been taking iron tablets for several years without having a checkup of her body iron status. After 22 phlebotomies over a period of 300 days, the iron excess was finally removed. |
The patient then shifts to an individual “maintenance treatment” with phlebotomy 2 - 4 times per year, in order to maintain the ferritin level at 50 - 100 µg/L and the transferrin saturation below 60%. Some especially frail elderly patients with co-morbidities do not tolerate the “standard phlebotomy regimen”; in these patients, a modified phlebotomy regimen may be required in the induction phase, where less than 450 - 500 mL blood is drawn and/or the intervals between phlebotomies may be extended. In the elderly, frail patients, a higher ferritin level of 200 - 400 µg/L may be accepted, in order to avoid anemia and discomfort due to the phlebotomies. In patients with difficult peripheral veins, ultrasound-guided venepuncture may be helpful.
In the induction phase the hemoglobin concentration should be measured prior to each phlebotomy, while ferritin and transferrin saturation are measured at every third to fourth phlebotomy. Plasma folate and plasma cobalamin should be checked at regular intervals and, if necessary, vitamin supplements with folic acid and vitamin B12 should be given.
In the maintenance phase, the patient should be checked with hemoglobin, ferritin and transferrin saturation every third to fourth month, and based on the results, a decision is made for phlebotomy. Patients should be followed lifelong on an out-patient basis.
In many countries, patients with preclinical hemochromatosis and patients with mild symptoms, but without chronic disease, are included in the blood donor corps and contribute extensively to cover the need for blood [67, 68]. In Denmark, there is no consensus on this issue, which is highly required in view of the facts that many blood donors actually have hemochromatosis without knowing it, and that many symptom-free patients with hemochromatosis become frustrated, because their blood is discarded and not used to help other patients.
In patients who do not tolerate phlebotomy, iron-chelating treatment with parenteral deferoxamine or oral deferiprone or oral deferasirox may be offered [69, 70]. There are ongoing clinical studies to assess the iron-chelating effect of deferasirox and K-deferasirox in patients with iron overload due to HFE-hemochromatosis. However, treatment with iron-chelating agents is significantly less effective than phlebotomy and may in some patients be associated with unpleasant side effects.
Several hepcidin agonists have been developed and are currently undergoing clinical trials. Results are pending; hopefully they will work and be approved for clinical use, which could be a major advance in the treatment of genetic hemochromatosis and other iron overload disorders [71].
| Prognosis | ▴Top |
A Danish study showed that patients with HFE-hemochromatosis, who are not treated or treated insufficiently, have a lower survival rate due to cirrhosis, HCC, cardiomyopathy and diabetes than patients receiving adequate iron-depletion treatment [72]. C282Y homozygous men with serum ferritin above 1,000 µg/L have a significantly higher prevalence of chronic fatigue, arthritis and liver disease than men without HFE-mutations [23]. The ferritin level at the time of diagnosis has prognostic significance, since patients with ferritin < 1,000 µg/L have a lower mortality rate than patients with ferritin > 2,000 µg/L [73, 74].
The earlier the diagnosis is made, the sooner treatment with phlebotomy is started; the lower the risk of developing organ dysfunction or organ damage is, the higher the survival rate will be. Patients in the preclinical stage and patients in the clinical stage without permanent organ dysfunction, such as liver cirrhosis, cardiomyopathy, pancreatic fibrosis and diabetes mellitus, will after adequate treatment have the same survival rate as the background population [75].
Although, at the time of diagnosis, organ damage, such as arthritis, liver cirrhosis, cardiomyopathy, pancreatic fibrosis and diabetes mellitus, is present, the patients must be treated with phlebotomy in order to prevent further deterioration of organ function. In most patients, treatment will lead to an improvement in organ functions (e.g. in patients with liver fibrosis, liver cirrhosis, cardiomyopathy and diabetes) [73-75], while arthralgias and arthritis often persist despite adequate iron-depletion treatment [42].
The production of gastric acid in the ventricle promotes the intestinal uptake of iron. Consequently, both antacids and proton pump inhibitors reduce iron uptake. In patients with hemochromatosis in the maintenance phase, administration of proton pump inhibitors may prolong the phlebotomy intervals [76, 77], which can be an advantage for some patients.
Patients with HFE-associated liver cirrhosis and hepatic insufficiency can be treated with liver transplantation, which “cures” the hemochromatosis, because the transplanted normal liver has the wild-type HFE-genotype and normal hepcidin production [78, 79]. Patients with life-threatening HFE-associated cardiomyopathy can benefit from cardiac transplantation or implantation of an artificial heart [79].
| Diet and Dietary Supplements | ▴Top |
In general, the diet of patients with hemochromatosis should follow the dietary guidelines of the Danish National Food Institute with regards to macro- and micronutrients, except for the iron content of the diet, which should be as low as possible. The absorption of both heme iron, mainly from meat, poultry and guts, and non-heme iron, mainly from vegetable food items, is increased in hemochromatosis, despite the presence of body iron overload. Besides the iron content per se in the foods, the balance between enhancers and inhibitors of iron absorption in the meal is important. Dietary adjustments can have an impact on how frequently the patient must be phlebotomized both during the induction and maintenance treatments but especially during the latter. However, it is up to the individual patient to decide to what extent he/she wants to change the diet in order to prolong the phlebotomy intervals. There are no controlled studies on the importance of the diet for the iron uptake in hemochromatosis, but presumably a carefully composed diet with a low iron content and being rich in inhibitors of iron absorption could significantly reduce the intestinal iron uptake [80].
Heme iron is absorbed 3 - 4 times better than non-heme iron, and in addition, meat and poultry contain the so-called “meat factors” that promote the absorption of non-heme iron. The meat intake, especially from red beef, which contains a high amount of heme iron, should therefore be limited. Vitamin C promotes iron absorption when consumed with the meal and vitamin supplements containing vitamin C must therefore be taken between the meals. Alcohol increases the uptake of iron from the duodenum significantly when taken with a meal [80]. Therefore, the alcohol intake should be limited and preferably consumed between meals and should not exceed the recommendations of the Danish National Board of Health, which is 14 units/week for men and 7 units/week for women.
Iron absorption is inhibited by phytic acid and phytates contained in cereals and bread and by polyphenols or tannins contained in tea and coffee. The content of polyphenols in one cup of black tea consumed with each meal has been shown to inhibit iron uptake and prolong the phlebotomy intervals [81]. Likewise, polyphenols are also available in tablets as dietary supplements. Calcium and magnesium in milk products also inhibit iron absorption and should be taken with meals.
As a consequence of the frequent phlebotomies in the induction phase, the body may be derived of protein and vitamins. Vitamin supplements may therefore be needed, especially folic acid and vitamin B12. Any kind of iron supplements including iron fortified foods must be avoided. Multivitamin-multimineral tablets without iron should be taken between meals. Calcium and vitamin D supplements for the prophylaxis and treatment of osteopenia or osteoporosis should be taken with meals. The frequent phlebotomies in the induction phase strongly stimulate iron absorption and many patients have an induction phase of 6 - 12 months or even longer. Consequently, dietary counseling and specific changes in the diet should be introduced early in the treatment phase. During the maintenance phase, the focus on the diet may have a positive impact on the patient’s sense of control of their own disease and can presumably reduce the number of phlebotomies.
| Family Screening | ▴Top |
When a person (proband) is diagnosed with HFE-hemochromatosis, the closest genetic relatives (i.e. the proband’s biological parents, siblings and children) should be examined with HFE-genotyping as to whether they have inherited the mutation(s). Due to the high frequency of C282Y and H63D heterozygosity in the Danish population, it is also recommended to examine the proband’s partner, when they have children. The examination of children may be postponed until it is convenient for both parents and children or until the children become mature.
| Population Screening | ▴Top |
Due to the high frequency of homozygosity and compound heterozygosity, and the significant long-term effects on health and quality of life as last shown in the UK Biobank Study [8], it should be considered whether screening of ethnic Danes for the HFE-hemochromatosis mutations should be implemented, as previously suggested in 1995 [29].
HFE-hemochromatosis meets the World Health Organization’s criteria for screening [82] and a study has shown that Danes have a very positive attitude towards screening for HFE-hemochromatosis [83]. An Australian study has demonstrated that several modalities of screening appear to be cost-effective for the society [84, 85].
Early diagnosis and treatment,which could be by blood donation, can save the society for considerable health-related costs [85]. Routine measurement of serum ferritin in blood donors in order to check their iron status prior to blood donation could identify many individuals in the preclinical stage. Important messages include: 1) HFE-hemochromatosis has been in the general health system for many years a disregarded genetic disorder/disease and the diagnosis is often made late in the clinical stage, implying considerable excess morbidity and mortality; 2) Among the five million ethnic Danes, at least 20,000 are homozygous and at least 500,000 are carriers of the HFE-mutations C282Y and H63D; 3) Diagnosis is obtained by blood analyses of serum ferritin, serum transferrin saturation and genetic testing for HFE-mutations; 4) Early diagnosis and treatment with phlebotomy prevent organ damage, increase quality of life and ensure normal survival rate; 5) Measurement of ferritin and transferrin saturation should be included the biochemical “routine analysis packages” used in the diagnostic evaluation of all patients presenting with health problems; 6) Persons having hemochromatosis in the preclinical and early clinical phase should be accepted as blood donors by the blood banks; and 7) Population screening for HFE-hemochromatosis appears to be cost-effective and should be considered.
In Denmark, all women are recommended to take iron supplements during pregnancy, including the approximately 400 women with HFE-mutations as well as the additional 20-30% of women who do not need extra iron. Among Danish women, more than 90% sooner or later become pregnant. Routine measurement of ferritin and transferrin saturation in pregnant women at their first check-up in the antenatal clinic (in Denmark at the general practitioner) in order to check iron status before prescribing iron supplements is recommended by the Danish Society of Obstetrics and Gynecology [86], but has still not been implemented by the Danish National Board of Health. Besides checking for iron deficiency, such a practice could identify women being genetically predisposed to HFE-hemochromatosis in the preclinical stage.
| Conclusions | ▴Top |
HFE-hemochromatosis is the most common genetic predisposition to disease in ethnic Danes as more than 20,000 individuals are homozygous for the C282Y mutation. The disease has a long preclinical stage with increasing iron accumulation in the body organs and approximately 30% of men develop clinical disease over time, while the clinical penetrance is low in premenopausal women but increases after the menopause.
Hemochromatosis is a multifaceted disease, in which the initial symptoms are fatigue, arthralgias, decreased libido, erectile dysfunction, signs of heart disease and diabetes mellitus. Later, organ dysfunction and organ damage occur in the form of liver cirrhosis, cardiomyopathy, pancreatic fibrosis and osteoporosis. The treatment consists of phlebotomy, which during the preclinical stage and in the early clinical stage will ensure a normal survival rate. Since the symptoms and organ damages are often irreversible, it is important that treatment is started early, namely before symptoms and organ dysfunction have developed.
Typically, many patients experience a long and confusing course in the health system and encounter various doctors and various specialties before the diagnosis is confirmed, often in an advanced clinical stage. This is evident in the UK Biobank Study [8] where only 22% of the men and 10% of the women aged 40 - 70 years had been diagnosed with hemochromatosis by the end of a 7-year follow-up period. It is therefore important that the specialties involved in the initial evaluation of patients presenting with symptoms, which are compatible with hemochromatosis (general practitioners, rheumatologists, endocrinologists, gastroenterologists, hepatologists, cardiologists) are aware of the disease and know how to make the diagnosis on a blood sample and then refer the patient to an appropriate clinic or hospital for further evaluation. Generally, the use of dietary iron supplements and treatment with iron tablets must always be based on the presence of iron deficiency, confirmed in a blood sample.
The Danish Hemochromatosis Association (Dansk Haemokromatose Forening) [87] is a patient-based organization, which was founded in 2012 and subsequently has been working to improve and spread the knowlegde about hereditary hemochromatosis both in the society and in the public health system.
Acknowledgments
None to declare.
Financial Disclosure
This study was supported by unrestricted grants from The Danish Haemochromatosis Association (Dansk Haemokromatose Forening) www.haemokromatose.dk and Pharmovital ApS, Rosenkaeret 11B, DK-2860 Soeborg, Denmark.
Conflict of Interest
All the authors declare that they have no conflict of interest.
Author Contributions
All four authors have contributed to the elaboration of the manuscript. NTM is the coordinator in the preparation of the manuscript and principal author. FVS, AEJ and KM are the co-authors of the manuscript.
| References | ▴Top |
- Milman NT. [Diagnosis and treatment of genetic haemochromatosis]. Ugeskr Laeger. 2013;175(16):1109-1112.
- Dansk Selskab for Gastroenterologi og Hepatologi (Danish Society for Gastroenterology and Hepatology). Hereditaer haemokromatose: Udredning, diagnostik og behandling (Hereditary hemochromatosis: evaluation, diagnosis and treatment) https://www.dsgh.dk/index.php/lever/hereditaer-hemokromatose-udredning-diagnostik-behandling. Accessed 1 June 2019.
- Milman NT, Schioedt FV, Junker AE, Magnussen K, Nathan T, Sandahl TD. Genetic HFE-haemochromatosis. Ugeskr Laeger. 2018;180(51):V09180619.
- Milman N, Pedersen P, Ovesen L, Melsen GV, Fenger K. Frequency of the C282Y and H63D mutations of the hemochromatosis gene (HFE) in 2501 ethnic Danes. Ann Hematol. 2004;83(10):654-657.
doi pubmed - Milman N, Pedersen P. Evidence that the Cys282Tyr mutation of the HFE gene originated from a population in Southern Scandinavia and spread with the Vikings. Clin Genet. 2003;64(1):36-47.
doi pubmed - Milman N, Koefoed P, Pedersen P, Nielsen FC, Eiberg H. Frequency of the HFE C282Y and H63D mutations in Danish patients with clinical haemochromatosis initially diagnosed by phenotypic methods. Eur J Haematol. 2003;71(6):403-407.
doi pubmed - Pedersen P, Milman N. Genetic screening for HFE hemochromatosis in 6,020 Danish men: penetrance of C282Y, H63D, and S65C variants. Ann Hematol. 2009;88(8):775-784.
doi pubmed - Pilling LC, Tamosauskaite J, Jones G, Wood AR, Jones L, Kuo CL, Kuchel GA, et al. Common conditions associated with hereditary haemochromatosis genetic variants: cohort study in UK Biobank. BMJ. 2019;364:k5222.
doi pubmed - European Association for the Study of the Liver. EASL clinical practice guidelines for HFE hemochromatosis. J Hepatol. 2010;53(1):3-22.
doi pubmed - Kew MC. Hepatic iron overload and hepatocellular carcinoma. Liver Cancer. 2014;3(1):31-40.
doi pubmed - Pietrangelo A. Hereditary hemochromatosis: pathogenesis, diagnosis, and treatment. Gastroenterology. 2010;139(2):393-408, e391-392.
doi pubmed - Milman N, Kirchhoff M. The influence of blood donation on iron stores assessed by serum ferritin and hemoglobin in a population survey of 1359 Danish women. Ann Hematol. 1991;63(1):27-32.
doi pubmed - Milman N, Kirchhoff M. Influence of blood donation on iron stores assessed by serum ferritin and haemoglobin in a population survey of 1433 Danish males. Eur J Haematol. 1991;47(2):134-139.
doi pubmed - Pedersen P, Milman N. Extrinsic factors modifying expressivity of the HFE variant C282Y, H63D, S65C phenotypes in 1,294 Danish men. Ann Hematol. 2009;88(10):957-965.
doi pubmed - Milman N. Iron status markers in hereditary haemochromatosis: distinction between individuals being homozygous and heterozygous for the haemochromatosis allele. Eur J Haematol. 1991;47(4):292-298.
doi pubmed - Milman N, Albeck MJ. Distinction between homozygous and heterozygous subjects with hereditary haemochromatosis using iron status markers and receiver operating characteristic (ROC) analysis. Eur J Clin Chem Clin Biochem. 1995;33(2):95-98.
doi pubmed - Adams PC, Barton JC. How I treat hemochromatosis. Blood. 2010;116(3):317-325.
doi pubmed - Milman N, Taylor CL, Merkel J, Brannon PM. Iron status in pregnant women and women of reproductive age in Europe. Am J Clin Nutr. 2017;106(Suppl 6):1655S-1662S.
doi pubmed - Milman N, Kirchhoff M, Jorgensen T. Iron status markers, serum ferritin and hemoglobin in 1359 Danish women in relation to menstruation, hormonal contraception, parity, and postmenopausal hormone treatment. Ann Hematol. 1992;65(2):96-102.
doi pubmed - Milman NT. Dietary Iron Intake in Women of Reproductive Age in Europe: A Review of 49 Studies from 29 Countries in the Period 1993-2015. J Nutr Metab. 2019;2019:7631306.
doi pubmed - Kelley M, Joshi N, Xie Y, Borgaonkar M. Iron overload is rare in patients homozygous for the H63D mutation. Can J Gastroenterol Hepatol. 2014;28(4):198-202.
doi pubmed - Gallego CJ, Burt A, Sundaresan AS, Ye Z, Shaw C, Crosslin DR, Crane PK, et al. Penetrance of Hemochromatosis in HFE Genotypes Resulting in p.Cys282Tyr and p.[Cys282Tyr];[His63Asp] in the eMERGE Network. Am J Hum Genet. 2015;97(4):512-520.
doi pubmed - Allen KJ, Gurrin LC, Constantine CC, Osborne NJ, Delatycki MB, Nicoll AJ, McLaren CE, et al. Iron-overload-related disease in HFE hereditary hemochromatosis. N Engl J Med. 2008;358(3):221-230.
doi pubmed - Gurrin LC, Bertalli NA, Dalton GW, Osborne NJ, Constantine CC, McLaren CE, English DR, et al. HFE C282Y/H63D compound heterozygotes are at low risk of hemochromatosis-related morbidity. Hepatology. 2009;50(1):94-101.
doi pubmed - Tamosauskaite J, Atkins JL, Pilling LC, Kuo CL, Kuchel GA, Ferrucci L, Melzer D. Hereditary Hemochromatosis Associations with Frailty, Sarcopenia and Chronic Pain: Evidence from 200,975 Older UK Biobank Participants. J Gerontol A Biol Sci Med Sci. 2019;74(3):337-342.
doi pubmed - Bassett ML, Halliday JW, Ferris RA, Powell LW. Diagnosis of hemochromatosis in young subjects: predictive accuracy of biochemical screening tests. Gastroenterology. 1984;87(3):628-633.
doi - Milman N. Hereditary haemochromatosis in Denmark 1950-1985. Clinical, biochemical and histological features in 179 patients and 13 preclinical cases. Dan Med Bull. 1991;38(4):385-393.
- Bell H, Berg JP, Undlien DE, Distante S, Raknerud N, Heier HE, Try K, et al. The clinical expression of hemochromatosis in Oslo, Norway. Excessive oral iron intake may lead to secondary hemochromatosis even in HFE C282Y mutation negative subjects. Scand J Gastroenterol. 2000;35(12):1301-1307.
doi pubmed - Groenbaek KE, Milman N, Skoedt V. [Preclinical hereditary hemochromatosis—is there an indication for preventive screening?]. Ugeskr Laeger. 1995;157(30):4249-4250.
- Sanchez-Luna SA, Brown KE. Clinical burden of liver disease from hemochromatosis at an academic medical center. Hepatol Commun. 2017;1(5):453-459.
doi pubmed - Poullis A, Moodie SJ, Ang L, Finlayson CJ, Levin GE, Maxwell JD. Routine transferrin saturation measurement in liver clinic patients increases detection of hereditary haemochromatosis. Ann Clin Biochem. 2003;40(Pt 5):521-527.
doi pubmed - Fracanzani AL, Conte D, Fraquelli M, Taioli E, Mattioli M, Losco A, Fargion S. Increased cancer risk in a cohort of 230 patients with hereditary hemochromatosis in comparison to matched control patients with non-iron-related chronic liver disease. Hepatology. 2001;33(3):647-651.
doi pubmed - Kew MC. Epidemiology of hepatocellular carcinoma. Toxicology. 2002;181-182:35-38.
doi - Kew MC. Prevention of hepatocellular carcinoma. Ann Hepatol. 2010;9(2):120-132.
doi - Elmberg M, Hultcrantz R, Ekbom A, Brandt L, Olsson S, Olsson R, Lindgren S, et al. Cancer risk in patients with hereditary hemochromatosis and in their first-degree relatives. Gastroenterology. 2003;125(6):1733-1741.
doi pubmed - Ellervik C, Tybjaerg-Hansen A, Nordestgaard BG. Risk of cancer by transferrin saturation levels and haemochromatosis genotype: population-based study and meta-analysis. J Intern Med. 2012;271(1):51-63.
doi pubmed - Fargion S, Valenti L, Fracanzani AL. Hemochromatosis gene (HFE) mutations and cancer risk: expanding the clinical manifestations of hereditary iron overload. Hepatology. 2010;51(4):1119-1121.
doi pubmed - Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL. Mitochondrial dysfunction in cardiac disease: ischemia—reperfusion, aging, and heart failure. J Mol Cell Cardiol. 2001;33(6):1065-1089.
doi pubmed - Aronow WS. Management of cardiac hemochromatosis. Arch Med Sci. 2018;14(3):560-568.
doi pubmed - Gaenzer H, Marschang P, Sturm W, Neumayr G, Vogel W, Patsch J, Weiss G. Association between increased iron stores and impaired endothelial function in patients with hereditary hemochromatosis. J Am Coll Cardiol. 2002;40(12):2189-2194.
doi - Skinner C, Kenmure AC. Haemochromatosis presenting as congestive cardiomyopathy and responding to venesection. Br Heart J. 1973;35(4):466-468.
doi pubmed - Harty LC, Lai D, Connor S, Dunne A, Ali M, Ryan J, O'Connell PG, et al. Prevalence and progress of joint symptoms in hereditary hemochromatosis and symptomatic response to venesection. J Clin Rheumatol. 2011;17(4):220-222.
doi pubmed - Husar-Memmer E, Stadlmayr A, Datz C, Zwerina J. HFE-related hemochromatosis: an update for the rheumatologist. Curr Rheumatol Rep. 2014;16(1):393.
doi pubmed - Kiely PD. Haemochromatosis arthropathy - a conundrum of the Celtic curse. J R Coll Physicians Edinb. 2018;48(3):233-238.
doi pubmed - Handzlik-Orlik G, Holecki M, Wilczynski K, Dulawa J. Osteoporosis in liver disease: pathogenesis and management. Ther Adv Endocrinol Metab. 2016;7(3):128-135.
doi pubmed - Bergeron C, Kovacs K. Pituitary siderosis. A histologic, immunocytologic, and ultrastructural study. Am J Pathol. 1978;93(2):295-309.
- Kelly TM, Edwards CQ, Meikle AW, Kushner JP. Hypogonadism in hemochromatosis: reversal with iron depletion. Ann Intern Med. 1984;101(5):629-632.
doi pubmed - Barton JC, Leiendecker-Foster C, Reboussin DM, Adams PC, Acton RT, Eckfeldt JH, Hemochromatosis, et al. Thyroid-stimulating hormone and free thyroxine levels in persons with HFE C282Y homozygosity, a common hemochromatosis genotype: the HEIRS study. Thyroid. 2008;18(8):831-838.
doi pubmed - Pappachan JM, Babu S, Krishnan B, Ravindran NC. Non-alcoholic Fatty Liver Disease: A Clinical Update. J Clin Transl Hepatol. 2017;5(4):384-393.
doi pubmed - Gandon Y, Olivie D, Guyader D, Aube C, Oberti F, Sebille V, Deugnier Y. Non-invasive assessment of hepatic iron stores by MRI. Lancet. 2004;363(9406):357-362.
doi - Legros L, Bardou-Jacquet E, Latournerie M, Guillygomarc'h A, Turlin B, Le Lan C, Desille Y, et al. Non-invasive assessment of liver fibrosis in C282Y homozygous HFE hemochromatosis. Liver Int. 2015;35(6):1731-1738.
doi pubmed - Adhoute X, Foucher J, Laharie D, Terrebonne E, Vergniol J, Castera L, Lovato B, et al. Diagnosis of liver fibrosis using FibroScan and other noninvasive methods in patients with hemochromatosis: a prospective study. Gastroenterol Clin Biol. 2008;32(2):180-187.
doi pubmed - Castera L, Foucher J, Bernard PH, Carvalho F, Allaix D, Merrouche W, Couzigou P, et al. Pitfalls of liver stiffness measurement: a 5-year prospective study of 13,369 examinations. Hepatology. 2010;51(3):828-835.
doi pubmed - Guyader D, Gandon Y, Deugnier Y, Jouanolle H, Loreal O, Simon M, Bourel M, et al. Evaluation of computed tomography in the assessment of liver iron overload. A study of 46 cases of idiopathic hemochromatosis. Gastroenterology. 1989;97(3):737-743.
doi - Malvarosa I, Massaroni C, Liguori C, Paul J, Beomonte Zobel B, Saccomandi P, Vogl TJ, et al. Estimation of liver iron concentration by dual energy CT images: influence of X-ray energy on sensitivity. Conf Proc IEEE Eng Med Biol Soc. 2014;2014:5129-5132.
doi pubmed - Sarigianni M, Liakos A, Vlachaki E, Paschos P, Athanasiadou E, Montori VM, Murad MH, et al. Accuracy of magnetic resonance imaging in diagnosis of liver iron overload: a systematic review and meta-analysis. Clin Gastroenterol Hepatol. 2015;13(1):55-63 e55.
doi pubmed - Assis RA, Kay FU, Conti FM, Campregher PV, Szarf G, Diniz MS, Rodrigues M, et al. The role of magnetic resonance imaging-T2* in the evaluation of iron overload early in hereditary hemochromatosis. A cross-sectional study with 159 patients. Am J Hematol. 2015;90(12):E220-221.
doi pubmed - Anderson LJ, Holden S, Davis B, Prescott E, Charrier CC, Bunce NH, Firmin DN, et al. Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. Eur Heart J. 2001;22(23):2171-2179.
doi pubmed - From AM, Maleszewski JJ, Rihal CS. Current status of endomyocardial biopsy. Mayo Clin Proc. 2011;86(11):1095-1102.
doi pubmed - Hahalis G, Manolis AS, Apostolopoulos D, Alexopoulos D, Vagenakis AG, Zoumbos NC. Right ventricular cardiomyopathy in beta-thalassaemia major. Eur Heart J. 2002;23(2):147-156.
doi pubmed - Dallos T, Sahinbegovic E, Aigner E, Axmann R, Schoniger-Hekele M, Karonitsch T, Stamm T, et al. Validation of a radiographic scoring system for haemochromatosis arthropathy. Ann Rheum Dis. 2010;69(12):2145-2151.
doi pubmed - Bazzocchi A, Ponti F, Albisinni U, Battista G, Guglielmi G. DXA: Technical aspects and application. Eur J Radiol. 2016;85(8):1481-1492.
doi pubmed - Powell LW, Seckington RC, Deugnier Y. Haemochromatosis. Lancet. 2016;388(10045):706-716.
doi - Adams P, Altes A, Brissot P, Butzeck B, Cabantchik I, Cancado R, Distante S, et al. Therapeutic recommendations in HFE hemochromatosis for p.Cys282Tyr (C282Y/C282Y) homozygous genotype. Hepatol Int. 2018;12(2):83-86.
doi pubmed - Ong SY, Gurrin LC, Dolling L, Dixon J, Nicoll AJ, Wolthuizen M, Wood EM, et al. Reduction of body iron in HFE-related haemochromatosis and moderate iron overload (Mi-Iron): a multicentre, participant-blinded, randomised controlled trial. Lancet Haematol. 2017;4(12):e607-e614.
doi - Goodnough LT. Erythropoietin and iron-restricted erythropoiesis. Exp Hematol. 2007;35(4 Suppl 1):167-172.
doi pubmed - Winters AC, Tremblay D, Arinsburg S, Mascarenhas J, Schiano TD. Reassessing the safety concerns of utilizing blood donations from patients with hemochromatosis. Hepatology. 2018;67(3):1150-1157.
doi pubmed - Bentley P, Bell B, Olynyk J. Therapeutic venesection at the Australian Red Cross Blood Service: impact of the High Ferritin Application on management of hereditary haemochromatosis. Aust Fam Physician. 2015;44(8):589-592.
- Fabio G, Minonzio F, Delbini P, Bianchi A, Cappellini MD. Reversal of cardiac complications by deferiprone and deferoxamine combination therapy in a patient affected by a severe type of juvenile hemochromatosis (JH). Blood. 2007;109(1):362-364.
doi pubmed - Binding A, Ward R, Tomlinson G, Kuo KHM. Deferiprone exerts a dose-dependent reduction of liver iron in adults with iron overload. Eur J Haematol. 2019;103(2):80-87.
doi pubmed - Casu C, Nemeth E, Rivella S. Hepcidin agonists as therapeutic tools. Blood. 2018;131(16):1790-1794.
doi pubmed - Milman N, Pedersen P, a Steig T, Byg KE, Graudal N, Fenger K. Clinically overt hereditary hemochromatosis in Denmark 1948-1985: epidemiology, factors of significance for long-term survival, and causes of death in 179 patients. Ann Hematol. 2001;80(12):737-744.
doi pubmed - Fracanzani AL, Fargion S, Romano R, Conte D, Piperno A, D'Alba R, Mandelli C, et al. Portal hypertension and iron depletion in patients with genetic hemochromatosis. Hepatology. 1995;22(4 Pt 1):1127-1131.
doi pubmed - Bardou-Jacquet E, Morcet J, Manet G, Laine F, Perrin M, Jouanolle AM, Guyader D, et al. Decreased cardiovascular and extrahepatic cancer-related mortality in treated patients with mild HFE hemochromatosis. J Hepatol. 2015;62(3):682-689.
doi pubmed - McDonnell SM, Preston BL, Jewell SA, Barton JC, Edwards CQ, Adams PC, Yip R. A survey of 2,851 patients with hemochromatosis: symptoms and response to treatment. Am J Med. 1999;106(6):619-624.
doi - Hutchinson C, Geissler CA, Powell JJ, Bomford A. Proton pump inhibitors suppress absorption of dietary non-haem iron in hereditary haemochromatosis. Gut. 2007;56(9):1291-1295.
doi pubmed - van Aerts RM, van Deursen CT, Koek GH. Proton pump inhibitors reduce the frequency of phlebotomy in patients with hereditary hemochromatosis. Clin Gastroenterol Hepatol. 2016;14(1):147-152.
doi pubmed - Bardou-Jacquet E, Philip J, Lorho R, Ropert M, Latournerie M, Houssel-Debry P, Guyader D, et al. Liver transplantation normalizes serum hepcidin level and cures iron metabolism alterations in HFE hemochromatosis. Hepatology. 2014;59(3):839-847.
doi pubmed - Robinson MR, Al-Kindi SG, Oliveira GH. Heart and heart-liver transplantation in patients with hemochromatosis. Int J Cardiol. 2017;244:226-228.
doi pubmed - Moretti D, van Doorn GM, Swinkels DW, Melse-Boonstra A. Relevance of dietary iron intake and bioavailability in the management of HFE hemochromatosis: a systematic review. Am J Clin Nutr. 2013;98(2):468-479.
doi pubmed - Kaltwasser JP, Werner E, Schalk K, Hansen C, Gottschalk R, Seidl C. Clinical trial on the effect of regular tea drinking on iron accumulation in genetic haemochromatosis. Gut. 1998;43(5):699-704.
doi pubmed - Burke W, Coughlin SS, Lee NC, Weed DL, Khoury MJ. Application of population screening principles to genetic screening for adult-onset conditions. Genet Test. 2001;5(3):201-211.
doi pubmed - Elsass P, Pedersen P, Husum K, Milman N. Assessment of the psychological effects of genetic screening for hereditary hemochromatosis. Ann Hematol. 2008;87(5):397-404.
doi pubmed - de Graaff B, Si L, Neil AL, Yee KC, Sanderson K, Gurrin LC, Palmer AJ. Population screening for hereditary haemochromatosis in Australia: construction and validation of a state-transition cost-effectiveness model. Pharmacoecon Open. 2017;1(1):37-51.
doi pubmed - de Graaff B, Neil A, Si L, Yee KC, Sanderson K, Gurrin L, Palmer AJ. Cost-effectiveness of different population screening strategies for hereditary haemochromatosis in Australia. Appl Health Econ Health Policy. 2017;15(4):521-534.
doi pubmed - Madsen LRF, Bulow NS, Tanvig M, Oldenburg A, Andersen LLT, Skorstengaard M, Petersen L, et al. Diagnostik og behandlingafjernmangeligraviditeten (Diagnostics and treatment of iron deficiency in pregnancy). Ugeskr Laeger. 2018;180:V03180210.
- Dansk Haemokromatose Forening (Danish Hemochromatosis Association). https://www.hemokromatose.dk/. Accessed July 1, 2019.
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Gastroenterology Research is published by Elmer Press Inc.