

| Gastroenterology Research, ISSN 1918-2805 print, 1918-2813 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Gastroenterol Res and Elmer Press Inc |
| Journal website https://www.gastrores.org |
Case Report
Volume 15, Number 5, October 2022, pages 268-277

An Unusual Solitary Fibrous Tumor of the Ischiorectal Region
Mahmoud R.A. Husseina, Abdullah Saad Alqahtanib, g, Mubarak Mohammed Al-Shraimc, Yahia Ibraheem Assirid, Feras O. Ahmedb, Mohammed Jalwi Korkomane, Ahmed Y. Al-Ameerf, Asmaa M. Ahmeda
aDepartment of Pathology, Faculty of Medicine, Assiut University, Assiut Governorate, Egypt
bDepartment of Surgery, Armed Forces Hospital Southern Region, Khamis Mushayt, Saudi Arabia
cDepartment of Pathology, College of Medicine, King Khalid University, Abha, Saudi Arabia
dDepartment of Radiology, College of Medicine, King Khalid University, Abha, Saudi Arabia
eDepartment of Surgery, University of Bisha, Bisha 61922, Saudi Arabia
fDepartment of Surgery, College of Medicine, University of Bisha, Bisha 61922, Saudi Arabia
gCorresponding Author: Abdullah Saad Alqahtani, Departments of General Surgery, Armed Forces Hospital Southern Region, Khamis Mushayt, Saudi Arabia
Manuscript submitted May 12, 2022, accepted August 8, 2022, published online October 19, 2022
Short title: Ischiorectal SFT
doi: https://doi.org/10.14740/gr1539
| Abstract | ▴Top |
Solitary fibrous tumors (SFTs) are rare fibroblastic/myofibroblastic proliferations that occur in a wide range of anatomical sites. These tumors have nonspecific clinical presentations often with unpredictable biological behavior. SFTs can be slow growing low-risk tumors or rapidly growing high-risk tumors. They show a wide variety of histological features and typically are characterized by NAB2-STAT6 fusion. SFTs of the ischiorectal fossa are rare, with few studies reported in the literature to date. Here, we report a 90-year-old male who had a road traffic accident in October 2018. A pelvic computed tomography (CT) revealed a mass measuring 3.5 × 2.5 cm in the right ischiorectal fossa. Histopathology of the CT-guided biopsies confirmed the diagnosis of low-grade SFT. No surgical intervention was needed since the patient was asymptomatic. In January 2022, a follow-up CT showed a gradual increase in tumor size (5 × 3.5 × 3 cm), but not infiltrating the surrounding structures. However, the patient complained of constipation, which warranted a surgical excision of the mass. Subsequently, immunohistological examination reconfirmed the diagnosis of low-risk SFT. Here, we discussed the clinicopathological features of the case and the relevant literature about pelvic SFTs. In conclusion, SFTs should be considered in the differential diagnosis of any ischiorectal mass. It is recommended that tissue samples be obtained, and immunohistology should be performed.
Keywords: SFT; Rectum; Tumor; Immunohistochemistry
| Introduction | ▴Top |
Historical aspects of solitary fibrous tumor
Solitary fibrous tumor (SFT) is rare mesenchymal tumor that Stout and Murray first described in 1931 as a pleural fibroma. Other names for SFT include benign mesothelioma, localized mesothelioma, pleural fibroma, subserosal fibroma, submesothelial fibroma, and subpleural fibroma. The incidence of this tumor is one new case per million people per year [1-3]. In 1942, Stout and Murray reviewed historical neoplasm called the “hemangiopericytomas” group. They found that although these neoplasms have seemingly similar histology in the form of profuse stag-horn vascular pattern, surrounded by connective tissue sheath, they did not share the same biological behavior. It has been challenging to diagnose these hemangiopericytomas, as this hemangiopericytoma-like pattern has also been observed in several other soft tissue tumors. The latter include mesenchymal chondrosarcomas, fibrosarcomas, and synovial sarcomas [1-7].
Hemangiopericytoma and SFT
After the discovery of NBA2-STAT6 gene fusion, World Health Organization (WHO) defined hemangiopericytoma and SFT as a single entity in 2016 [4-11].
Clinical locations of SFTs
SFT occurs most frequently in the pleura but has been described in several anatomic sites, including the viscera and soft tissues, especially the deep soft tissues. The thorax (lung, mediastinum, and diaphragm) is the most common extra-pleural site. SFTs can occur at intraperitoneal, retroperitoneal, or pelvic locations in the abdomen, which is the second most common extra-pleural site for SFTs [12-14]. Other rare anatomic sites that are affected by SFTs include the head and neck region, extremities, and meninges [11, 14-16]. However, the sites of predilection for SFTs are the pleura, peritoneum, and meninges [17]. WHO classification (2020) of tumors considered SFT as a neoplasm with fibroblastic/myofibroblastic differentiation that rarely metastasizes. SFTs are sometimes classified as intermediate malignant tumors [18].
The clinical features of the pelvic SFTs
SFTs of the pelvic area is very rare. In addition to the sigmoid, rectum (serosa) and mesorectum, they can be found in the prostate, urinary bladder, spinal sacral canal, perineal area, and ischiorectal and ischioanal fossae [14-16, 19-28]. There have been only a few reported cases of SFTs involving the ischiorectal fossa in the literature [15, 16, 19, 21, 22, 25, 26, 28].
SFTs of the pelvic region have no sex predilection, and the peak incidence is between 40 and 70 years [18]. The SFTs in the pelvis usually involve deep tissue rather than superficial. SFTs in the pelvis are generally asymptomatic until they become large enough to produce mass effects on the surrounding tissues. There are no specific clinical features of pelvic SFTs [15, 16, 19, 21, 22, 25, 26, 28]. They include abdominal pain [28], difficulty in urinating, urinary retention [27], numbness and weakness in lower extremities due to nerve compression, constipation, and dysuria [28, 29]. A summary of some previous cases is presented in Table 1 [14-16, 19-28, 30-32].
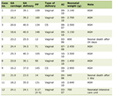 Click to view | Table 1. Previous Studies About Pelvic Solitary Fibrous Tumor |
The paraneoplastic syndromes and the SFTs
SFTs may rarely be associated with paraneoplastic syndromes, most commonly non-islet cell hypoglycemia with the production of insulin-like growth factors (IGFs), specifically IGF-II. Most of these tumors are malignant; therefore, the presence of non-islet cell hypoglycemia is considered a poor prognostic factor [33, 34]. Rarely, SFTs may also be associated with other paraneoplastic syndromes like Pierre-Marie-Bamberger syndrome [35], Doege-Potter syndrome [36], cerebellar degeneration [37], and hypertrophic osteoarthropathy [38].
The radiological features of the SFTs
The diagnostic approach of pelvic SFTs is similar to other soft tissue tumors at varied anatomical sites. The diagnosis of SFT is often incidental on radiography or CT [39], with appearance being nonspecific [39]. Imaging studies include plain radiographs that typically reveal a well-defined mass. Contrast-enhanced computed tomography (CT) usually shows a well-defined, hypervascular mass lesion.
SFT can be hypodense or hyperdense with respect to muscle. The attenuation of SFT depends on the content of the collagen bundles. The densely collagenized SFTs are usually hyperdense. Conversely, the hypodense SFTs usually have fewer collagen contents [39]. The SFTs are usually hypoechoic on ultrasonographic examination but occasionally may have a heterogeneous appearance [39]. Alternatively, on magnetic resonance imaging (MRI), these tumors are isointense on T1-weighted images and can be variable on T2-weighted images [40]. Benign SFTs have low-grade activity (hypometabolic state), whereas their malignant counterparts usually have hypermetabolic and homogeneous activity on fluorodeoxyglucose-positron emission tomography (FDG-PET) [41].
The gross and histological features of the pelvic SFTs
SFTs are typically more than 1.0 cm in diameter (size ranging from 1.0 to 40 cm) [42, 43]. They are typically well-defined, can have a smooth surface or be lobulated, may or may not have a capsule. Cut sections of these masses are usually yellow or grayish white. Cystic changes or necrosis is rare [44].
SFTs in the pelvis are characterized histologically by a spindle-to-ovoid-shaped proliferations of fibroblasts/myofibroblasts associated with staghorn-like vasculature. Some SFTs may have hemangiopericytoma-like or storiform pattern or can be patternless. Tumor cells consist of monomorphic or mildly atypical spindle-shaped to ovoid-shaped cells with minimal cytoplasm, arranged in undulating, straight, curved, or patternless arrangements [26, 43, 45]. Tumor cells are embedded in a variable cellular fibrous stroma which are separated by bands of hyalinized, ropy collagen. Some tumors have poorly cellular, densely collagenized stroma, while others have an abundant cellular stroma [11, 18]. Some tumors are homogenously cellular, while others have a cellular zone alternating with hypocellular or keloid-like areas. They are accompanied by hemangiopericytoma-like vessels [11, 18].
The grade of SFTs depends upon several histological features including the mitotic activity, necrosis and nuclear pleomorphism. The degree of mitotic activity may range from the complete absence or scarcity of mitotic activity to mitotically active stromal cells with some atypical mitotic figures. This feature is crucial in the risk stratification of SFTs. Moreover, the degree of nuclear pleomorphism, and necrosis are also important histological features for risk stratification of these tumors [11, 46].
Variants of the SFTs
SFTs have several histological variants [11]. The giant cell rich SFTs (formerly known as giant cell-angiofibroma) are common in the head and neck region and tend to be indolent. The differentiated SFTs (also known as anaplastic SFTs) are the most aggressive variants and contain areas of high-grade sarcoma encompassing heterologous elements such as bone-forming sarcoma or rhabdomyosarcoma [47, 48]. Fat-forming SFTs contain mature adipose tissue and have an indolent course similar to giant cell rich SFTs. Alternatively, this variant may contain lipoblast (lipomatous SFTs) and present with malignant behavior [47, 49].
The immunohistochemical and ultrastructural features of the SFTs
Immunohistochemically, the neoplastic cells in SFTs are usually strongly reactive to CD34 and STAT6. Although CD34 is a conventional marker in SFT, it can be expressed by other soft tissue tumors and therefore, lack specificity. Alternatively, STAT6 is considered the most sensitive and specific marker for SFTs. The tumors are also variably reactive to other markers, including CD99, BCL-2, EMA, and nuclear β-catenin [28, 29, 50]. The tumor cells of SFT are negative for SOX10, desmin, CD31, pancytokeratin (AE1/AE3), and S-100 [11]. An ultrastructural description of SFTs is that they are composed of cells that are fibroblast-like and have a well-developed rough endoplasmic reticulum. These cells are seen amid collagen fibers [51]. Some SFTs may also have a myxoid stroma [47].
Risk stratification of the SFTs
The behavior of SFTs has been difficult to predict. Our knowledge about the biological behavior and malignant potential of SFTs is rudimentary. Nonetheless, available case reports indicate that most of these tumors are indolent and slowly growing [44]. However, SFTs can metastasize in 5-25% of cases [46]. England et al proposed five criteria for judging malignant change in SFT. They include necrosis, mitotic activity (more than four mitotic figures per 10 high-power fields (HPFs)), high cellularity, pleomorphism, and hemorrhage [52].
The conventional sarcoma grading systems have proved to be poorly applicable to SFT. Also, the validity of the current sarcoma staging systems for traditional sarcomas has not been tested for these tumors. SFTs with sarcomatous changes usually behave aggressively and can metastasize like other high-grade sarcomas. Alternatively, it is difficult to predict the biological behavior of tumors lacking overt sarcomatous changes [53]. Based on the risk stratification, SFTs include low aggressive, highly aggressive, and dedifferentiated SFT. Out of the several risk classification models, only two of them have been validated. One model is based on age, size, mitotic count, and tumor necrosis and distributes the patients into three different risk categories. The other risk model estimates the individual risk for local and metastatic recurrence [11, 46].
The genetic features of the SFTs
NGFI-A binding (NAB) proteins can repress or activate transcription induced by some members of the EGR (early growth response) family of transactivators [54]. The STAT6 is a transcription factor. It is a member of STAT family of proteins [55]. The members of this family transmit signals from a receptor complex to the nucleus. They, therefore, activate gene expression. STAT6 is also activated by growth factors and cytokines such as interleukin-4 and interleukin-13. The transcription of NAB2-STAT6 genes occurs in the opposite direction. The fusion product results from an inversion at the 12q13 locus. The resultant NAB2-STAT6 fusion protein converts NAB2 from transcriptional repressor to activator. This leads to constitutive expression of the early growth response 1 (EGR1) target genes such as IGF2, FGFR1 [10, 56-59].
The somatic fusions of the two genes, located at chromosomal region 12q13, namely NGFI-A-binding protein 2 (NAB2) and STAT6, have been recently considered as the tumor-initiating events in SFTs [60]. STAT6 nuclear reactivity in SFTs has diagnostic sensitivity and specificity [58, 59]. The most common NAB2-STAT6 fusion variants in these tumors are NAB2ex4-STAT6ex2, NAB2ex6-STAT6ex16, and NAB2ex6-STAT6ex17. The NAB2ex4-fused SFTs represent a distinct from non-NAB2ex4-fused counterparts in several clinical and pathological aspects [61].
The treatment strategies of the SFTs
Given our rudimentary knowledge about SFTs, there are no clearly defined treatment protocols. Surgery (tumorectomy with wide negative surgical margin) is the mainstay. Surgical excision may be the only treatment necessary for SFTs of the pelvic region with no or low recurrence rate. Radiotherapy is not generally recommended after surgical excision as most of these tumors are indolent or low risk. Chemotherapy using certain drugs (such as bevacizumab, sunitinib, pazopanib, and sorafenib) that target the vascular endothelial growth factor and other tyrosine kinase signaling pathways are sometimes used. These pathways interfere with the blood supply to the tumor and are used to stop the progression of the tumor [62-65]. Patients with malignant SFTs or tumors with positive margins or unresectable or recurrent tumors may benefit from radiation therapy [66].
| Case Report | ▴Top |
A 90-year-old male, otherwise healthy, had a road traffic accident in October 2018. A CT scan was requested as a routine workup of trauma assessment. It revealed an incidental finding of asymptomatic round, well-defined soft tissue density with variable enhancement in the right ischiorectal fossa measuring 3.5 × 2.5 cm. Following this, MRI revealed a well-defined mass lesion of abnormal signal intensity measuring 3.5 × 2.5 cm in the right ischiorectal fossa, inseparable from the pelvic sidewall muscles laterally. The mass was in close contact with the rectum medially and superiorly. It was seen separable from but displacing the pelvic floor muscles. The mass lesion displayed mixed intermediate and hyperintense signals on the T2-weighted image. These findings were impressive of a neoplastic process. A summary of these findings is shown in Figure 1.
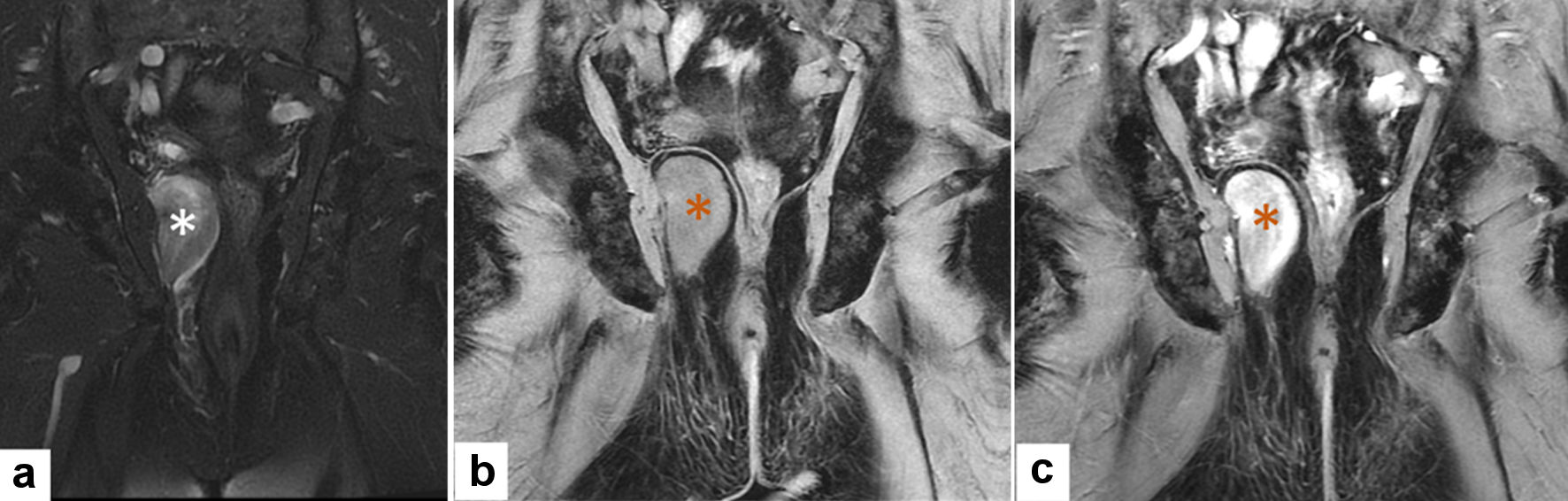 Click for large image | Figure 1. Radiological MRI features of solitary fibrous tumor (MRI: October 2018). (a) Coronal T2WI with fat saturation shows a well-defined heterogeneous predominantly low signal intensity right ischiorectal fossa ice-cone shaped mass (white asterisk). (b, c) Pre and post contrast-enhanced coronal T1WI with fat suppression showing homogenous lesion isointense to muscles in pre-contract and avidly enhancing following contrast administration (orange asterisk). MRI: magnetic resonance imaging. |
CT-guided biopsy was taken, and the histological examination revealed a spindle-cell-shaped neoplasm composed of patternless proliferation of short spindly cells with mitotically inactive nuclei, inconspicuous nucleoli, and pink cytoplasm with some collagen fibril network. The proliferating cells were admixed with small blood vessels (Fig. 2). The tumor cells were positive for CD34, BCL2, CD99, and STAT6. They were negative for desmin, S100, and pancytokeratin (AE1/AE3), supporting the diagnosis of SFT. The patient was offered an excisional biopsy, but he refused.
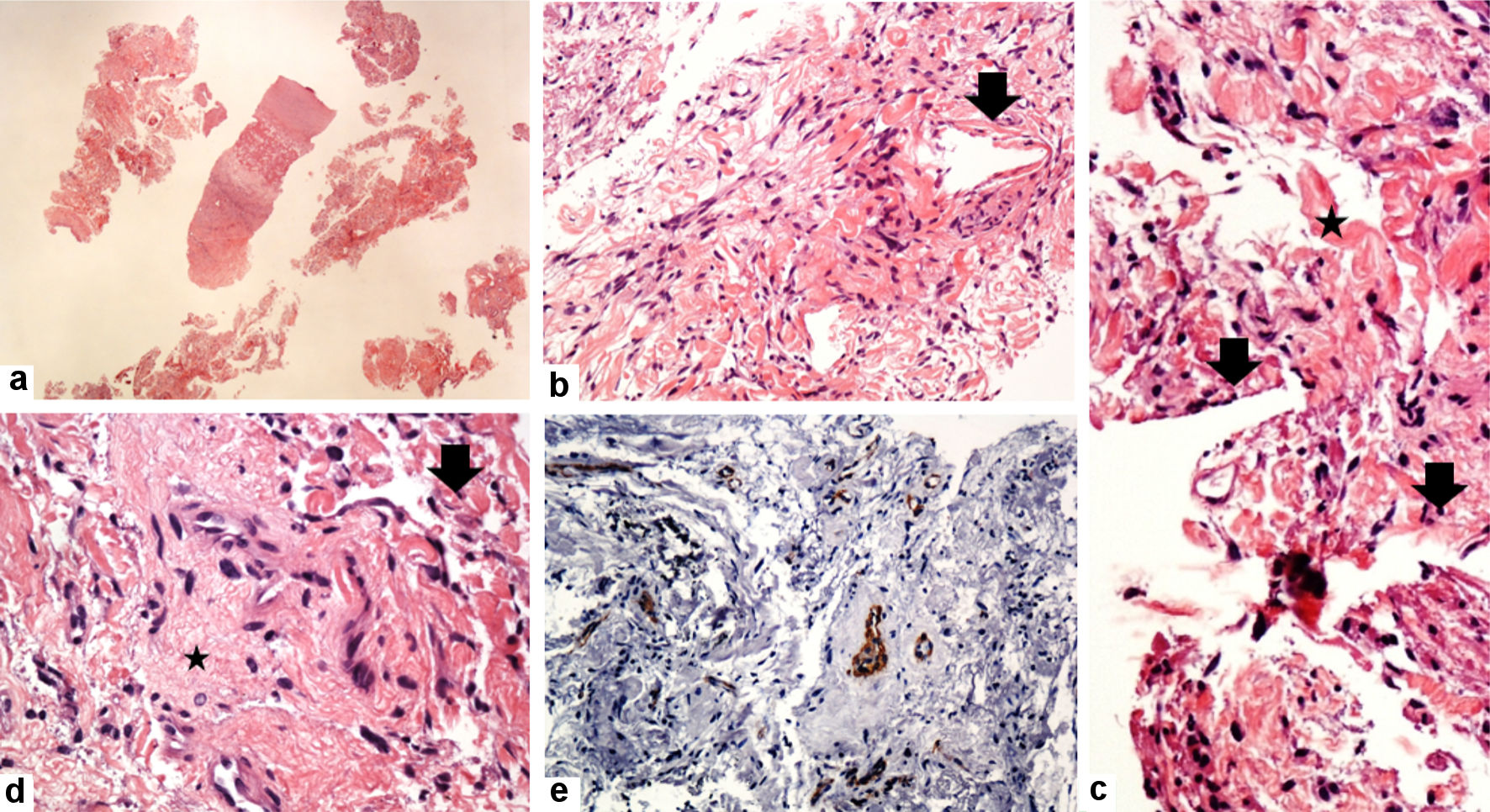 Click for large image | Figure 2. Histological features of solitary fibrous tumors (true cut needle biopsy: October 2018). (a-d) The tumor consists of a haphazard proliferation of short spindle to ovoid cells with banal-looking oval to spindle nuclei, pale eosinophilic cytoplasm, and some collagenized stroma (star), and the hemangiopericytoma-like vascular structures (arrow). The tumor cells are negative for SMA (e) (original magnifications: (a) × 20; (b) × 200; (c) × 400; (d) × 200; and (e) × 200). |
More than 3 years later (January 2022), the patient came back with newly developed vague symptoms related to the mass lesion (constipation). A follow-up imaging showed only a mild increase in the lesion size. Radiological studies revealed well-defined heterogeneous predominantly low-signal right ischiorectal fossa ice-cone-shaped mass (Figs. 3, 4). The patient agreed to surgical intervention (excision of the mass). The tumor was below the pelvic floor and therefore abdominal approach is not an option. The patient was put in lithotomy position, and under general anesthesia, a longitudinal incision was made just lateral to the site of the external sphincter muscles. The surgical dissection was extended upward till the site of the mass. The lesion was soft, movable, and compressible. The mass could not be grasped and taken out without applying a digital transrectal pushing the mass outward. The mass was released, delivered out, and excised completely. The wound was closed in a layer, and a Penrose drain was used to drain the cavity and prevent fluid collection and infection.
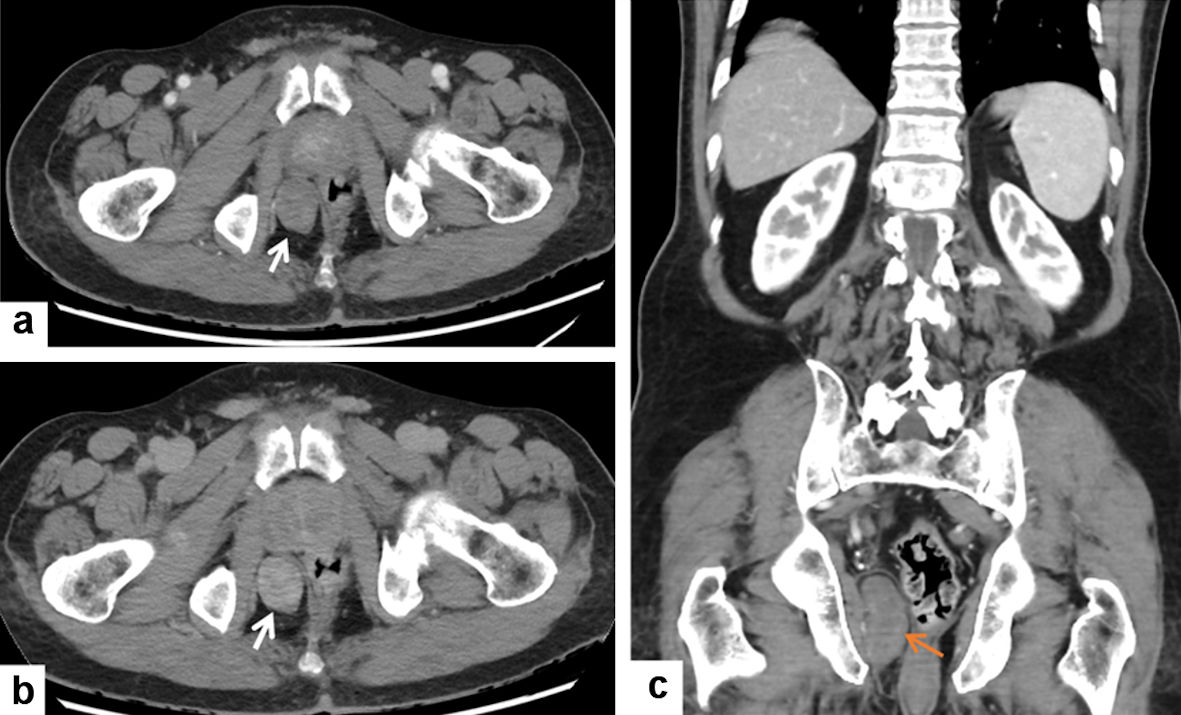 Click for large image | Figure 3. Radiological CT features of solitary fibrous tumor (CT: January 2022). (a, b) Contrast-enhanced CT of the pelvis shows a well-defined right ischiorectal fossa oval-shaped mass (white arrow) with progressive contrast enhancement on delayed CT (b). (c) Contrast-enhanced CT coronal reformat shows a well-defined elongated right ischiorectal fossa mass (orange arrow). CT: computed tomography. |
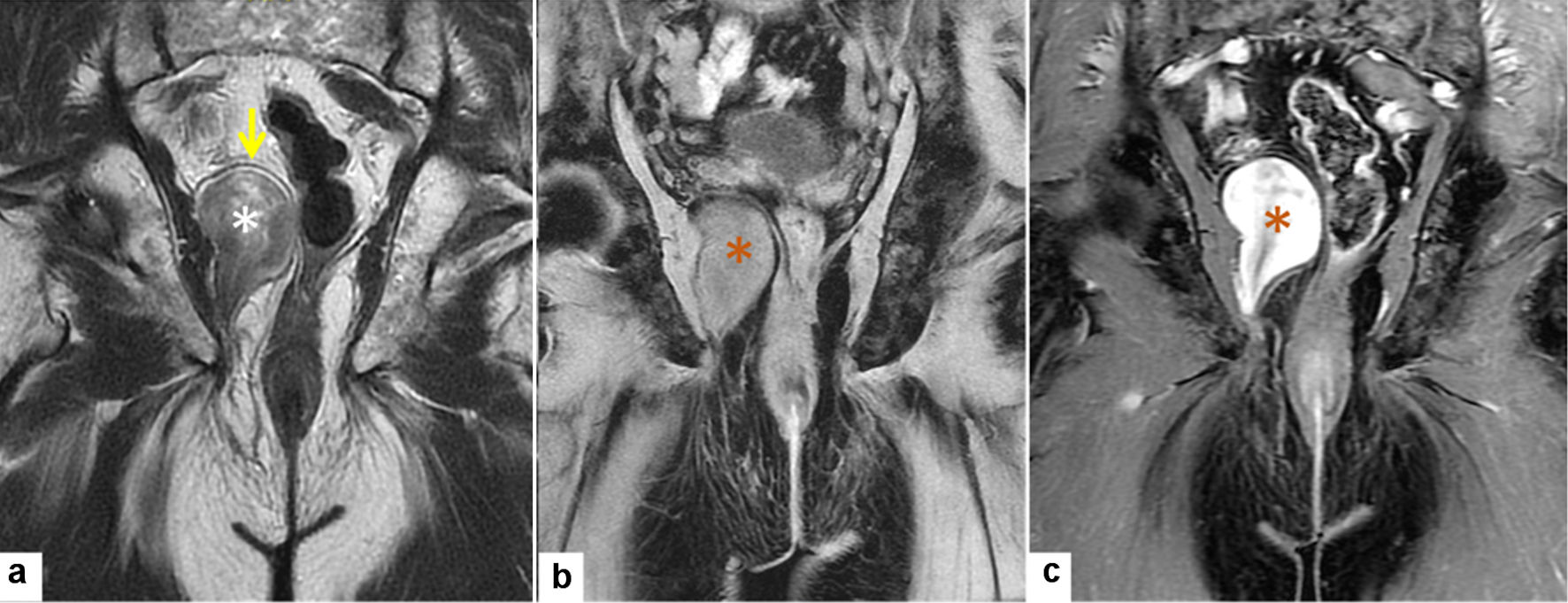 Click for large image | Figure 4. Radiological MRI features of solitary fibrous tumor (MRI: January 2022). (a) Coronal T2WI shows right well-defined heterogeneous predominantly low signal right ischiorectal fossa ice cone-shaped mass (white asterisk) upward displacing the right levator ani muscle (yellow arrow) without invasion. (b, c) Pre- and post-contrast-enhanced coronal T1WI with fat suppression show homogenous lesion isointense to muscles in pre-contract and avidly enhancing following contrast administration (orange asterisk). MRI: magnetic resonance imaging. |
Gross examination revealed a well-defined, thinly encapsulated mass measuring 5 × 3.5 × 3 cm with a pink cut section and firm consistency. Histological examination revealed haphazard proliferation of spindly and oval mitotically inactive cells with bland short spindly and oval nuclei, pale eosinophilic cytoplasm, and variable amounts of the collagenous stroma. The tumor was cellular, and the mitotic count was 0/10 HPFs. There was no evidence of necrosis or hemorrhage (Fig. 5). The resection margins were free. The postoperative recovery was uneventful, and the drain was removed 2 weeks later without any complications. According to the four-variable model for risk stratification, the tumor was considered as low risk (score of 3) [43].
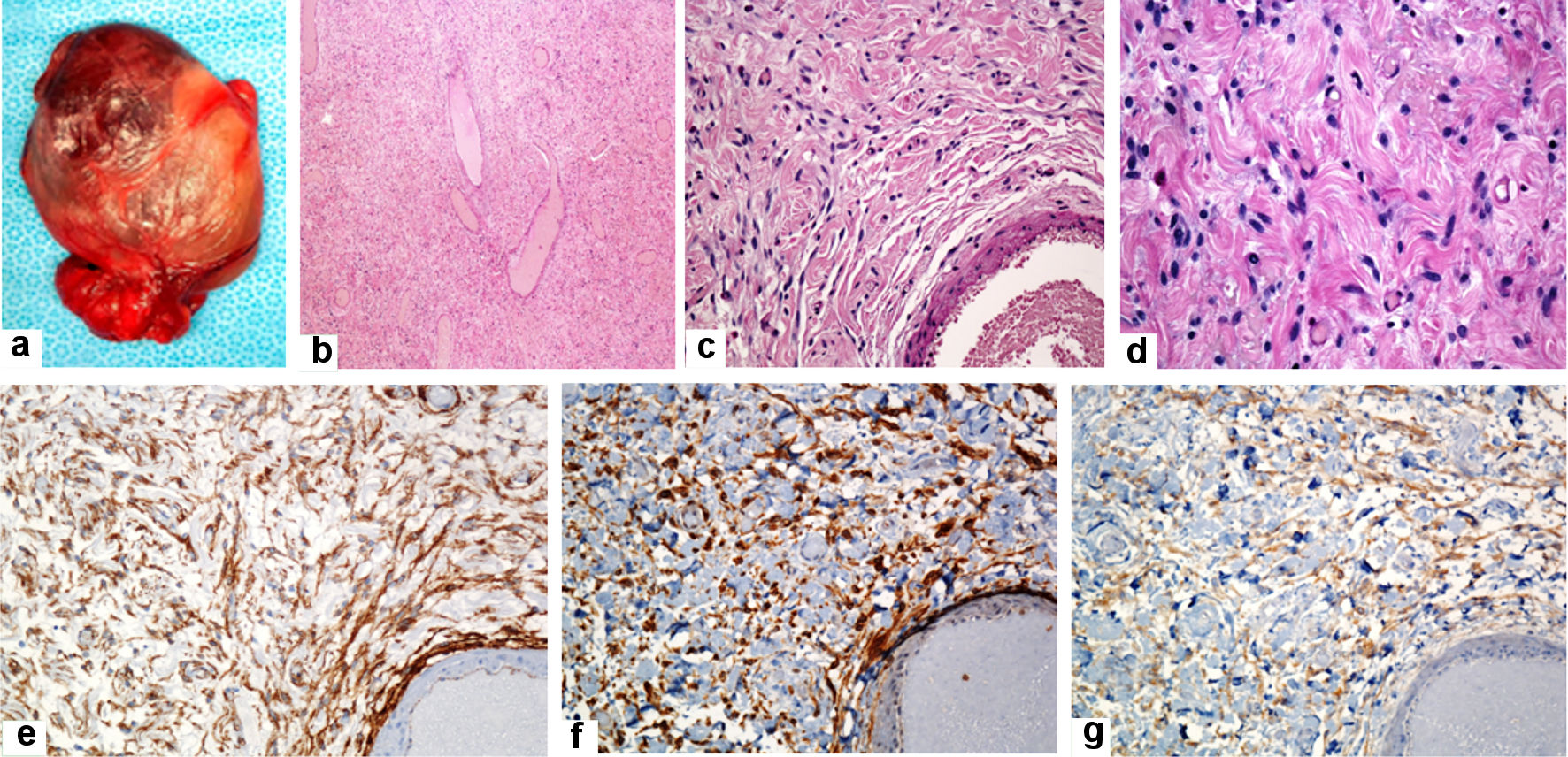 Click for large image | Figure 5. Gross and histological features of the solitary fibrous tumor (excisional biopsy: January 2022). Gross examination of the mass reveals well-defined, thinly encapsulated mass measuring about 5 × 3.5 × 3 cm (a). Histologic sections reveal cellular spindle cell neoplasm in a collagenous stroma with variable-sized blood vessels having stag-horn vascular morphology (b). The neoplastic cells are ovoid, short, or fusiform spindle-shaped cells with indistinct cell borders, bland looking nuclei, and indistinct nucleoli. They are arranged in short, poorly defined bundles, haphazardly fashion, and patternless pattern. The cells are streamed among the dermal collagens. No mitotic activity was seen. No significant nuclear atypia or necrosis was seen (c, d). The tumor cells are diffusely and strongly positive for CD34, BCL2, and CD99 (e, f, and g, respectively) and are negative for desmin, S100, and pancytokeratin (AE1/AE3) (original magnifications: (b) × 40, (c) × 200, (d) × 400, (e) × 200, (f) × 200, and (g) × 200). |
| Discussion | ▴Top |
Given that SFTs of the pelvic region are rare neoplasms, much of the literature has emerged from the occasional case reports. Therefore, our knowledge about this entity is not only limited but also fragmented. We pursued this study to improve our understanding of these neoplasms. To achieve our goal, we presented a literature review about pelvic SFTs. Also, herein, we presented a case of ischiorectal SFT in an elderly male patient. The characteristic immunohistological profiles of the tumor and the nonspecific clinical manifestations of the case presented here concur with previous studies [14-16, 19-28]. Nassif et al [44] presented a literature review of five reported cases of anorectal SFTs [14, 19, 24, 67, 68]. The tumors occurred in four males and one female. The size of the tumors ranged from 7 cm to 13 cm. The surgical resection with a clear margin was the mainstay of treatment in all cases. The patient did not receive chemotherapy or radiotherapy. None of the patients received adjuvant chemotherapy radiation [44].
In the case reported herein, there was a slow increase in the size of the tumor over a period of more than 3 years. This indicates the indolent nature of the tumor. Moreover, the assessment of risk stratification suggests that the tumor belongs to the low-risk group following the scheme proposed by Demicco et al and Bhat et al [30, 46]. In 2012, Demicco et al initially reported a three-variable scheme (age, tumor size, and mitosis) on the risk stratification for SFT [46]. In 2017, the authors revised their scheme by incorporating tumor necrosis (necrosis representing 10% or more of the tumor) as a new risk factor to their previously reported variables (patient age, tumor size, and mitotic activity). According to this revised scheme, the revised risk stratification model for SFT comprised of low risk (0 - 3), intermediate-risk (4 - 5), and high risk (6 - 7) groups. The authors validated their revised scheme in 79 patients with primary non-meningeal SFTs. Most of the patients (66%) were at low risk and had no metastasis at 10 years. Some patients (24%) were scored as an intermediate risk with a 10% risk of metastasis at 10 years. The high risk included 10% of the patients with a 73% risk of metastasis at 5 years [46]. According to this risk stratification, the case presented here is considered as the low-risk tumor. The slow growth and lack of invasion of the surrounding ischiorectal structures support the indolent behavior of the tumor.
The histological differential diagnosis of the ischiorectal region SFT reported herein includes other soft tissue tumors with the hemangiopericytic-like vascular pattern, including synovial sarcoma (negative for STAT6 and CD34 and has SS18-SSX gene fusions) [69], dermatofibrosarcoma protuberans (positive for CD34 but negative for STAT6, and has OLIA1-PDGFB gene fusions) [70], deep fibrous histiocytoma (negative for STAT6) [71], and liposarcoma (MDM2 gene amplification by fluorescence in situ hybridisation (FISH)) [72]. The other tumors in that should be separated from SFTs include mesenchymal chondrosarcoma (chondroid component, negative for STAT6 and the presence of HEY1-NCOA2 gene fusions) [73, 74], myopericytoma (positive for smooth muscle actin and negative for STAT6) [75], gastrointestinal stromal tumor (positive for CD34, DOG1, and CD117 but negative for STAT6) [76], and mammary type myofibroblastoma (positive for desmin and ER but negative for STAT6) [77].
To conclude, SFTs are mesenchymal neoplasms with fibroblastic/myofibroblastic differentiation and unpredictable biological behavior. The physicians should be aware of this entity whenever presented with any ischiorectal mass lesion.
Learning points
Although SFTs were first reported in the pleura (localized fibrous mesothelioma), they were subsequently reported in several anatomical sites.
Pelvic SFT, including the tumors of the ischiorectal and ischioanal fossae, perineal region, sigmoid (serosa), rectum (serosa), mesorectum, prostate, urinary bladder, sacral spinal canal, obturator area, are rare.
There are no sex predilections in SFTs.
SFTs most commonly occur in the age range between 20 -70 years.
The somatic fusions of NAB2 and STAT6 genes are the tumor-initiating events in SFT.
The clinical presentations of the pelvic SFTs are nonspecific, and they include constipation, dysuria, bleeding per rectum, pain in the lower extremities associated with numbness, and muscle weakness.
SFTs are histologically composed of spindle cells and a hemangiopericytoma-like vasculature. The spindled cells can show variable cytologic atypia, pleomorphism, and mitotic figures/10HPFs, but no hemorrhage or necrosis. Immunohistochemically, the tumor cells show strong and diffuse positivity for CD34 and STAT6. The neoplastic cells are negative for desmin, smooth muscle actin, inhibin, and CD117.
The hemangiopericytoma-like vasculature and the salient histological features of SFTs.
The histological variants of SFTs include the fat-forming SFTs, the giant cell-rich SFTs, and the dedifferentiated SFTs.
STAT6 nuclear reactivity in SFTs has diagnostic sensitivity and specificity [58, 59].
The features of malignancy in SFTs include increased cellularity, mitosis, and necrosis.
The behavior of SFTs has been difficult to predict.
Risk-stratification of SFTs is based on the patient’s age, size of the tumor, mitotic activity, and tumor necrosis.
Based on the risk stratification, SFTs include low-risk, intermediate-risk, and high-risk tumors.
Surgical excision (tumorectomy) is the standard management in most cases of pelvic SFTs.
The beneficial roles of adjuvant chemotherapy or radiotherapy is still unclear.
Acknowledgments
None to declare.
Financial Disclosure
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sector.
Conflict of Interest
None to declare.
Informed Consent
The informed consent was obtained from the patient.
Author Contributions
All authors certify that he or she has equally participated sufficiently in the intellectual content, the analysis of data and the writing up of the manuscript. Each author has reviewed the final version of the manuscript and approved it for publication.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Hussein MR, Nassar MI, Kamel NA, Osman ME, Georguis MN. Analysis of fibronectin expression in the bilharzial granulomas and of laminin in the transformed urothelium in schistosoma haematobium infested patients. Cancer Biol Ther. 2005;4(6):676-678.
doi pubmed - Klemperer P, Coleman BR. Primary neoplasms of the pleura. A report of five cases. Am J Ind Med. 1992;22(1):1-31.
doi pubmed - Stiller CA, Trama A, Serraino D, Rossi S, Navarro C, Chirlaque MD, Casali PG, et al. Descriptive epidemiology of sarcomas in Europe: report from the RARECARE project. Eur J Cancer. 2013;49(3):684-695.
doi pubmed - Stout AP. Mesotheliomas of the pleura and peritoneum. J Tn State Med Assoc. 1951;44(10):409-411.
- Stout AP. Tumors of the pleura. Harlem Hosp Bull. 1952;5(2):54-57.
- Stout AP, Himadi GM. Solitary (localized) mesothelioma of the pleura. Ann Surg. 1951;133(1):50-64.
doi pubmed - Stout AP, Murray MR. Hemangiopericytoma: a vascular tumor featuring Zimmermann's pericytes. Ann Surg. 1942;116(1):26-33.
doi pubmed - Soutelo J, Moldes S, Martin A, Lutfi R, Leal Reyna M. [Hypoglycemia induced by a solitary fibrous tumor of the lung or Doege-Potter syndrome: Report of one case]. Rev Med Chil. 2016;144(1):129-133.
doi pubmed - Davanzo B, Emerson RE, Lisy M, Koniaris LG, Kays JK. Solitary fibrous tumor. Transl Gastroenterol Hepatol. 2018;3:94.
doi pubmed - Smrke A, Thway K, P HH, Jones RL, Hayes AJ. Solitary fibrous tumor: molecular hallmarks and treatment for a rare sarcoma. Future Oncol. 2021;17(27):3627-3636.
doi pubmed - Martin-Broto J, Mondaza-Hernandez JL, Moura DS, Hindi N. A comprehensive review on solitary fibrous tumor: new insights for new horizons. Cancers (Basel). 2021;13(12):2913.
doi pubmed - Vallat-Decouvelaere AV, Dry SM, Fletcher CD. Atypical and malignant solitary fibrous tumors in extrathoracic locations: evidence of their comparability to intra-thoracic tumors. Am J Surg Pathol. 1998;22(12):1501-1511.
doi pubmed - Gold JS, Antonescu CR, Hajdu C, Ferrone CR, Hussain M, Lewis JJ, Brennan MF, et al. Clinicopathologic correlates of solitary fibrous tumors. Cancer. 2002;94(4):1057-1068.
doi pubmed - Dudkiewicz M, Deschenes JL, Bloom C, Gordon PH. Solitary fibrous tumor of the ischioanal fossa: report of a case and review of the literature. Dis Colon Rectum. 2004;47(4):535-537.
doi pubmed - Morikawa K, Takenaga S, Masuda K, Kano A, Igarashi T, Ojiri H, Ueda K, et al. A rare solitary fibrous tumor in the ischiorectal fossa: a case report. Surg Case Rep. 2018;4(1):126.
doi pubmed - Paramythiotis D, Moysidis M, Kourtidis L, Karakatsanis A, Poulios C, Michalopoulos A. Perianal solitary fibrous tumor in a rare anatomical presentation: a case report and literature review. Am J Case Rep. 2021;22:e929742.
doi pubmed - Natteru P, Ramachandran Nair L, Luzardo G, Shaikh N. Meningeal hemangiopericytoma presenting as pure Gerstmann syndrome: A Double Rarity. Cureus. 2021;13(6):e15863.
doi pubmed - Sugita S, Segawa K, Kikuchi N, Takenami T, Kido T, Emori M, Akiyama Y, et al. Prognostic usefulness of a modified risk model for solitary fibrous tumor that includes the Ki-67 labeling index. World J Surg Oncol. 2022;20(1):29.
doi pubmed - Yoshida R, Takada H, Iwamoto S, Uedono Y, Kawanishi H, Yoshioka K, Nakane Y, et al. A solitary fibrous tumor in the perianal region with a 13-year follow-up: report of a case. Surg Today. 1999;29(7):642-645.
doi pubmed - Yuza K, Sakata J, Nagaro H, Ando T, Hirose Y, Miura K, Takizawa K, et al. A giant pelvic solitary fibrous tumor with Doege-Potter syndrome successfully treated with transcatheter arterial embolization followed by surgical resection: a case report. Surg Case Rep. 2020;6(1):299.
doi pubmed - Soda H, Kainuma O, Yamamoto H, Nagata M, Takiguchi N, Ikeda A, Cho A, et al. Giant intrapelvic solitary fibrous tumor arising from mesorectum. Clin J Gastroenterol. 2010;3(3):136-139.
doi pubmed - Ishihara H, Omae K, Iizuka J, Kobayashi H, Fukuda I, Kondo T, Hizuka N, et al. Late recurrence of a malignant hypoglycemia-inducing pelvic solitary fibrous tumor secreting high-molecular-weight insulin-like growth factor-II: A case report with protein analysis. Oncol Lett. 2016;12(1):479-484.
doi pubmed - Ishikawa T, Kawabata G, Terakawa T, Kamidono S, Fujisawa M. Solitary fibrous tumor in the pelvic space. Urol Res. 2004;32(1):49-50.
doi pubmed - Joe BN, Bolaris M, Horvai A, Yeh BM, Coakley FV, Meng MV. Solitary fibrous tumor of the male pelvis: findings at CT with histopathologic correlation. Clin Imaging. 2008;32(5):403-406.
doi pubmed - Kawamura J, Tani M, Kida Y, Sumida K, Ogawa R, Kawasoe J, Yazawa T, et al. Successful laparoscopic treatment of a giant solitary fibrous tumor of the mesorectum: A case report and literature review. Asian J Endosc Surg. 2017;10(1):51-54.
doi pubmed - Matsui Y, Hamada M, Sumiyama F, Kobayashi T, Matsumi Y, Miki H, Ishida M, et al. Two cases of primary solitary fibrous tumor in the pelvis resected using laparoscopic surgery. Int J Surg Case Rep. 2020;71:58-65.
doi pubmed - Sano T, Nishiyama H, Kanematsu A, Takahashi T, Nakamura E, Kamoto T, Mikami Y, et al. [Solitary fibrous tumor in the pelvic space: a case report]. Hinyokika Kiyo. 2007;53(12):897-901.
- Pata F, Orsini V, Lucisano AM, Pafundi DP, Sacco R. Solitary fibrous tumor of the pelvis: an uncommon soft-tissue tumor. A case report. Ann Ital Chir. 2010;81(6):457-460.
- Ronchi A, La Mantia E, Gigantino V, Perdona S, De Sio M, Facchini G, Franco R, et al. A rare case of malignant solitary fibrous tumor in prostate with review of the literature. Diagn Pathol. 2017;12(1):50.
doi pubmed - Bhat A, Layfield LJ, Tewari SO, Gaballah AH, Davis R, Wu Z. Solitary fibrous tumor of the ischioanal fossa-a multidisciplinary approach to management with radiologic-pathologic correlation. Radiol Case Rep. 2018;13(2):468-474.
doi pubmed - Furuta T, Nakai Y, Gonoi W, Kurokawa R, Okimoto N, Sakamoto N, Fukuchi H, et al. Fat-forming solitary fibrous tumor of the sacrum: A case report and literature review. Radiol Case Rep. 2021;16(7):1874-1877.
doi pubmed - Bratton L, Salloum R, Cao W, Huber AR. Solitary fibrous tumor of the sigmoid colon masquerading as an adnexal neoplasm. Case Rep Pathol. 2016;2016:4182026.
doi pubmed - Herrmann BL, Saller B, Kiess W, Morgenroth K, Drochner K, Schroder T, Mann K. Primary malignant fibrous histiocytoma of the lung: IGF-II producing tumor induces fasting hypoglycemia. Exp Clin Endocrinol Diabetes. 2000;108(8):515-518.
doi pubmed - Bruzzone A, Varaldo M, Ferrarazzo C, Tunesi G, Mencoboni M. Solitary fibrous tumor. Rare Tumors. 2010;2(4):e64.
doi pubmed - Boyer-Duck E, Dajer-Fadel WL, Hernandez-Arenas LA, Macias-Morales MP, Rodriguez-Gomez A, Romo-Aguirre C. Pierre-Marie-Bamberger syndrome and solitary fibrous tumor: a rare association. Asian Cardiovasc Thorac Ann. 2018;26(2):154-157.
doi pubmed - Chen S, Zheng Y, Chen L, Yi Q. A broad ligament solitary fibrous tumor with Doege-Potter syndrome. Medicine (Baltimore). 2018;97(39):e12564.
doi pubmed - Karki A, Yang J, Chauhan S. Paraneoplastic syndrome associate with solitary fibrous tumor of pleura. Lung India. 2018;35(3):245-247.
doi pubmed - Fridlington J, Weaver J, Kelly B, Kelly E. Secondary hypertrophic osteoarthropathy associated with solitary fibrous tumor of the lung. J Am Acad Dermatol. 2007;57(5 Suppl):S106-110.
doi pubmed - Rosado-de-Christenson ML, Abbott GF, McAdams HP, Franks TJ, Galvin JR. From the archives of the AFIP: Localized fibrous tumor of the pleura. Radiographics. 2003;23(3):759-783.
doi pubmed - Weon YC, Kim EY, Kim HJ, Byun HS, Park K, Kim JH. Intracranial solitary fibrous tumors: imaging findings in 6 consecutive patients. AJNR Am J Neuroradiol. 2007;28(8):1466-1469.
doi pubmed - Ginat DT, Bokhari A, Bhatt S, Dogra V. Imaging features of solitary fibrous tumors. AJR Am J Roentgenol. 2011;196(3):487-495.
doi pubmed - Brunnemann RB, Ro JY, Ordonez NG, Mooney J, El-Naggar AK, Ayala AG. Extrapleural solitary fibrous tumor: a clinicopathologic study of 24 cases. Mod Pathol. 1999;12(11):1034-1042.
- Demicco EG, Park MS, Araujo DM, Fox PS, Bassett RL, Pollock RE, Lazar AJ, et al. Solitary fibrous tumor: a clinicopathological study of 110 cases and proposed risk assessment model. Mod Pathol. 2012;25(9):1298-1306.
doi pubmed - Nassif MO, Trabulsi NH, Bullard Dunn KM, Nahal A, Meguerditchian AN. Soft tissue tumors of the anorectum: rare, complex and misunderstood. J Gastrointest Oncol. 2013;4(1):82-94.
- Feasel P, Al-Ibraheemi A, Fritchie K, Zreik RT, Wang WL, Demicco E, Saeb-Lima M, et al. Superficial Solitary Fibrous Tumor: A Series of 26 Cases. Am J Surg Pathol. 2018;42(6):778-785.
doi pubmed - Demicco EG, Wagner MJ, Maki RG, Gupta V, Iofin I, Lazar AJ, Wang WL. Risk assessment in solitary fibrous tumors: validation and refinement of a risk stratification model. Mod Pathol. 2017;30(10):1433-1442.
doi pubmed - Furusato E, Valenzuela IA, Fanburg-Smith JC, Auerbach A, Furusato B, Cameron JD, Rushing EJ. Orbital solitary fibrous tumor: encompassing terminology for hemangiopericytoma, giant cell angiofibroma, and fibrous histiocytoma of the orbit: reappraisal of 41 cases. Hum Pathol. 2011;42(1):120-128.
doi pubmed - Thway K, Hayes A, Ieremia E, Fisher C. Heterologous osteosarcomatous and rhabdomyosarcomatous elements in dedifferentiated solitary fibrous tumor: further support for the concept of dedifferentiation in solitary fibrous tumor. Ann Diagn Pathol. 2013;17(5):457-463.
doi pubmed - Lee JC, Fletcher CD. Malignant fat-forming solitary fibrous tumor (so-called "lipomatous hemangiopericytoma"): clinicopathologic analysis of 14 cases. Am J Surg Pathol. 2011;35(8):1177-1185.
doi pubmed - Herawi M, Epstein JI. Solitary fibrous tumor on needle biopsy and transurethral resection of the prostate: a clinicopathologic study of 13 cases. Am J Surg Pathol. 2007;31(6):870-876.
doi pubmed - Hirano D, Mashiko A, Murata Y, Satoh K, Ichinose T, Takahashi S, Jike T, et al. A case of solitary fibrous tumor of the kidney: an immunohistochemical and ultrastructural study with a review of the literature. Med Mol Morphol. 2009;42(4):239-244.
doi pubmed - England DM, Hochholzer L, McCarthy MJ. Localized benign and malignant fibrous tumors of the pleura. A clinicopathologic review of 223 cases. Am J Surg Pathol. 1989;13(8):640-658.
doi pubmed - Mosquera JM, Fletcher CD. Expanding the spectrum of malignant progression in solitary fibrous tumors: a study of 8 cases with a discrete anaplastic component—is this dedifferentiated SFT? Am J Surg Pathol. 2009;33(9):1314-1321.
doi pubmed - Kim J, Kang SM, Oh SY, Lee HJ, Lee I, Hwang JC, Hong SH. NGFI-A binding protein 2 promotes EGF-dependent HNSCC cell invasion. Cancers (Basel). 2019;11(3):315.
doi pubmed - Shahmarvand N, Nagy A, Shahryari J, Ohgami RS. Mutations in the signal transducer and activator of transcription family of genes in cancer. Cancer Sci. 2018;109(4):926-933.
doi pubmed - Kuo YS, Tang YB, Lu TY, Wu HC, Lin CT. IGFBP-6 plays a role as an oncosuppressor gene in NPC pathogenesis through regulating EGR-1 expression. J Pathol. 2010;222(3):299-309.
doi pubmed - Mohajeri A, Tayebwa J, Collin A, Nilsson J, Magnusson L, von Steyern FV, Brosjo O, et al. Comprehensive genetic analysis identifies a pathognomonic NAB2/STAT6 fusion gene, nonrandom secondary genomic imbalances, and a characteristic gene expression profile in solitary fibrous tumor. Genes Chromosomes Cancer. 2013;52(10):873-886.
doi pubmed - Doyle LA, Tao D, Marino-Enriquez A. STAT6 is amplified in a subset of dedifferentiated liposarcoma. Mod Pathol. 2014;27(9):1231-1237.
doi pubmed - Doyle LA, Vivero M, Fletcher CD, Mertens F, Hornick JL. Nuclear expression of STAT6 distinguishes solitary fibrous tumor from histologic mimics. Mod Pathol. 2014;27(3):390-395.
doi pubmed - Bieg M, Moskalev EA, Will R, Hebele S, Schwarzbach M, Schmeck S, Hohenberger P, et al. Gene expression in solitary fibrous tumors (SFTs) correlates with anatomic localization and NAB2-STAT6 gene fusion variants. Am J Pathol. 2021;191(4):602-617.
doi pubmed - Tai HC, Chuang IC, Chen TC, Li CF, Huang SC, Kao YC, Lin PC, et al. NAB2-STAT6 fusion types account for clinicopathological variations in solitary fibrous tumors. Mod Pathol. 2015;28(10):1324-1335.
doi pubmed - Park MS, Patel SR, Ludwig JA, Trent JC, Conrad CA, Lazar AJ, Wang WL, et al. Activity of temozolomide and bevacizumab in the treatment of locally advanced, recurrent, and metastatic hemangiopericytoma and malignant solitary fibrous tumor. Cancer. 2011;117(21):4939-4947.
doi pubmed - Stacchiotti S, Negri T, Libertini M, Palassini E, Marrari A, De Troia B, Gronchi A, et al. Sunitinib malate in solitary fibrous tumor (SFT). Ann Oncol. 2012;23(12):3171-3179.
doi pubmed - Park MS, Ravi V, Conley A, Patel SR, Trent JC, Lev DC, Lazar AJ, et al. The role of chemotherapy in advanced solitary fibrous tumors: a retrospective analysis. Clin Sarcoma Res. 2013;3(1):7.
doi pubmed - Stacchiotti S, Tortoreto M, Baldi GG, Grignani G, Toss A, Badalamenti G, Cominetti D, et al. Preclinical and clinical evidence of activity of pazopanib in solitary fibrous tumour. Eur J Cancer. 2014;50(17):3021-3028.
doi pubmed - Robinson LA. Solitary fibrous tumor of the pleura. Cancer Control. 2006;13(4):264-269.
doi pubmed - Hasegawa T, Matsuno Y, Shimoda T, Hasegawa F, Sano T, Hirohashi S. Extrathoracic solitary fibrous tumors: their histological variability and potentially aggressive behavior. Hum Pathol. 1999;30(12):1464-1473.
doi - Mourra N, Lewin M, Sautet A, Parc R, Flejou JF. Epithelioid solitary fibrous tumor in the ischioanal fossa. Virchows Arch. 2005;446(6):674-676.
doi pubmed - Amary MF, Berisha F, Bernardi Fdel C, Herbert A, James M, Reis-Filho JS, Fisher C, et al. Detection of SS18-SSX fusion transcripts in formalin-fixed paraffin-embedded neoplasms: analysis of conventional RT-PCR, qRT-PCR and dual color FISH as diagnostic tools for synovial sarcoma. Mod Pathol. 2007;20(4):482-496.
doi pubmed - Silverman JS, Tamsen A. A cutaneous case of giant cell angiofibroma occurring with dermatofibrosarcoma protuberans and showing bimodal CD34+ fibroblastic and FXIIIa+ histiocytic immunophenotype. J Cutan Pathol. 1998;25(5):265-270.
doi pubmed - Piombino E, Broggi G, Barbareschi M, Castorina S, Parenti R, Bartoloni G, Salvatorelli L, et al. Wilms' Tumor 1 (WT1): A novel immunomarker of dermatofibrosarcoma protuberans-an immunohistochemical study on a series of 114 cases of bland-looking mesenchymal spindle cell lesions of the dermis/subcutaneous tissues. Cancers (Basel). 2021;13(2):252.
doi pubmed - Kaneko K, Sawada S, Satake C, Kondo K, Izumi T, Tanaka M, Imai J, et al. Extraordinarily long-inactive solitary fibrous tumor transformed to produce big insulin-like growth factor-2, leading to hypoglycemia and rapid liposarcoma growth: a case report. BMC Endocr Disord. 2020;20(1):148.
doi pubmed - He R, Patel RM, Alkan S, Hammadeh R, Weiss SW, Goldblum JR, Venkataraman G, et al. Immunostaining for SYT protein discriminates synovial sarcoma from other soft tissue tumors: analysis of 146 cases. Mod Pathol. 2007;20(5):522-528.
doi pubmed - Schuetze SM, Bolejack V, Choy E, Ganjoo KN, Staddon AP, Chow WA, Tawbi HA, et al. Phase 2 study of dasatinib in patients with alveolar soft part sarcoma, chondrosarcoma, chordoma, epithelioid sarcoma, or solitary fibrous tumor. Cancer. 2017;123(1):90-97.
doi pubmed - Edgecombe A, Peterson RA, Shamji FM, Commons S, Sekhon H, Gomes MM. Myopericytoma: a pleural-based spindle cell neoplasm off the beaten path. Int J Surg Pathol. 2011;19(2):247-251.
doi pubmed - Shidham VB, Chivukula M, Gupta D, Rao RN, Komorowski R. Immunohistochemical comparison of gastrointestinal stromal tumor and solitary fibrous tumor. Arch Pathol Lab Med. 2002;126(10):1189-1192.
doi pubmed - Fritchie KJ, Carver P, Sun Y, Batiouchko G, Billings SD, Rubin BP, Tubbs RR, et al. Solitary fibrous tumor: is there a molecular relationship with cellular angiofibroma, spindle cell lipoma, and mammary-type myofibroblastoma? Am J Clin Pathol. 2012;137(6):963-970.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Gastroenterology Research is published by Elmer Press Inc.