

| Gastroenterology Research, ISSN 1918-2805 print, 1918-2813 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Gastroenterol Res and Elmer Press Inc |
| Journal website https://www.gastrores.org |
Original Article
Volume 14, Number 1, February 2021, pages 13-20
Characterization of Chronic Gastritis in Lynch Syndrome Patients With Gastric Adenocarcinoma
David Saulinoa, Rong Chena, Kai Wanga, Minqian Shenb, Xuefeng Zhangb, Maria Westerhoffc, Jerome Chengc, Jingmei Lind, Xuchen Zhange, Michael Feelya, Xiuli Liua, f
aDepartment of Pathology, Immunology and Laboratory Medicine, University of Florida, Gainesville, FL 32610, USA
bDepartment of Anatomic Pathology, Cleveland Clinic, Cleveland, OH 44195, USA
cDepartment of Pathology, University of Michigan, Ann Arbor, MI 48109, USA
dDepartment of Pathology, Indiana University, Indianapolis, IN 46202, USA
eDepartment of Pathology, Yale University, New Haven, CT 06510, USA
fCorresponding Author: Xiuli Liu, Department of Pathology, Immunology and Laboratory Medicine, University of Florida, P.O. Box 100275, 1600 SW Archer Road, Gainesville, FL 32610, USA
Manuscript submitted December 29, 2020, accepted January 15, 2021, published online February 19, 2021
Short title: Characterization of CG in LS Patients With GA
doi: https://doi.org/10.14740/gr1359
| Abstract | ▴Top |
Background: Gastric cancer is one of the Lynch syndrome (LS)-associated malignancies. Previous studies have suggested that LS patients with gastric cancer also had chronic atrophic gastritis in the background mucosa, but further histologic characterization was not attempted. This study aims to understand the histologic features of background chronic gastritis in LS patients with gastric adenocarcinoma.
Methods: Eleven LS-associated gastric cancer cases were collected from five institutions. Demographics and clinical features were retrieved by review of medical charts. Pathological material was reviewed for tumor location and histologic type. In addition, non-neoplastic gastric mucosa was assessed for inflammation (chronic and active), atrophy, intestinal metaplasia (IM) in the antrum and body, as well as pyloric gland metaplasia and enterochromaffin-like (ECL) cell hyperplasia in the body.
Results: Eleven LS patients with gastric cancer (four male and seven female) with a mean age of 63 years (range: 23 - 83) were included. Ten (90.9%) had personal cancer histories; however none of the patients had family history of gastric cancer. Eight (72.7%) patients underwent gastrectomy and three had endoscopic resection. Nine (81.8%) patients had tumor in the fundus and/or body and two had tumor present in the antrum. Seven (63.6%) cases were intestinal type or mixed type carcinoma, and the remaining four were signet ring cell carcinoma. Eight (of 11, 72.7%) patients had chronic gastritis, five (45.4%) had atrophy, and four (36.3%) had intestinal metaplasia. Four of five patients with both antrum and body mucosa available for evaluation (80%), demonstrated body-predominant chronic gastritis. Four patients had germline MLH1 alterations and all of these patients had chronic gastritis, including one Helicobacter pylori (H. pylori) gastritis and three H. pylori-negative gastritis.
Conclusions: None of LS patients with gastric cancer in our cohort had a family history of gastric cancer. Gastric adenocarcinomas in LS patients were primarily located in the fundus and/or body. Two-thirds of these tumors were of intestinal type and had a background chronic, H. pylori-negative gastritis. These results support a chronic atrophic gastritis with intestinal metaplasia-dysplasia-carcinoma sequence in LS-related gastric tumorigenesis, particularly in MLH1-mutated LS patients.
Keywords: Lynch syndrome; Gastric adenocarcinoma; Chronic gastritis; Atrophy; Intestinal metaplasia; H. pylori; MLH1
| Introduction | ▴Top |
Lynch syndrome (LS), also known as hereditary nonpolyposis colorectal cancer (HNPCC), is one of the most common inherited cancer syndromes and confers increased risk for cancers of many different body sites [1]. The stomach is regarded as the second most common extracolonic site that LS-related tumors can manifest [1]. One previous large study reported that 1.6% of LS patients developed gastric cancer [2]. These tumors are most often associated with germline MLH1 or MSH2 alterations, and family history of gastric cancer has been found to be a poor indicator for individual risk [2].
Gastric tumorigenesis in sporadic cases includes sequential changes that begin with chronic gastritis, subsequent atrophy/intestinal metaplasia, dysplasia and eventually to carcinoma in most gastric adenocarcinomas [3]. The chronic gastritis described in this setting is most often Helicobacter pylori (H. pylori)-related gastritis, with autoimmune gastritis being the cause in the minority of cases.
Previous reports have suggested that chronic atrophic gastritis is also present in the background of LS-related gastric cancers. A pattern of chronic atrophic gastritis limited to the antrum was reported in one kindred from southern Italy that had an excess of gastric carcinoma [4]. While the H. pylori gastritis rate has been reported to be approximately 20-27% in LS patients who developed adenocarcinoma [5, 6], which is similar to LS patients without gastric cancer [7] and LS patients’ family members without mismatch repair (MMR) gene mutations [8]. Additionally, a high prevalence of “autoimmune” gastritis was observed in MSH2-mutated LS patients who developed gastric adenocarcinoma [7]. However, this “autoimmune” gastritis in LS patients who developed gastric adenocarcinoma has not been well characterized histologically. It remains unknown if this type of “autoimmune” gastritis occurs in LS patients carrying mutations in other MMR genes such as MLH1, MSH6, and PMS2. Answers to these questions would shed light on the gastric tumorigenesis in LS patients and thus further risk stratify LS patients for gastric cancer surveillance.
In this study, we identified 11 patients with LS associated gastric cancer via a multicentric collaborative effort from five institutions, retrieved basic demographics and clinical features, reviewed the pathologic material for tumor characteristics and examined the background gastric mucosa changes. We also compared the findings in the background gastric mucosa in our cohort of LS patients with gastric cancer with the previously published data in LS patients without gastric adenocarcinoma [8].
| Materials and Methods | ▴Top |
This study was approved by the Institutional Research Boards of the participating institutions; and the study was conducted in compliance with the ethical standards of the responsible institution on human subjects as well as with the Helsinki Declaration.
Patients
A total of 11 cases of LS-associated gastric adenocarcinoma spanning the years 2010 - 2019 were retrieved from the pathology archives of five institutions including: the University of Michigan (four cases), Cleveland Clinic (three cases), the University of Indiana (one case), Yale University (one case), and the University of Florida (two cases) using search terms of “Lynch syndrome”, “hereditary non-polyposis colorectal cancer” and “gastric carcinoma” or a combination of the above. Inclusion criteria were: 1) a documented clinical diagnosis of LS based on personal and family cancer history and/or genetically confirmed germline mutations in MMR (MLH1, MSH2, MSH6, or PMS2) genes; 2) histological documentation of gastric adenocarcinoma; and 3) available adjacent/background gastric mucosa tissue for histological assessment according to previously published criteria [9, 10].
The information on basic demographics, reason for endoscopy if applicable (surveillance or symptoms), clinical laboratory test results including autoantibodies for autoimmune gastritis and/or ancillary testing for H. pylori, and personal or family tumor history were retrieved via chart review.
Pathologic material including surgical pathology reports and slides from each case was re-reviewed by an experienced gastrointestinal (GI) pathologist from the participating institutions. The tumor features included in this study were location (fundus, body, and antrum), type (intestinal vs. diffuse vs. mixed), depth of invasion, nodal metastases, or distant metastases. Immunohistochemical stains for MMR proteins were performed at the time of initial workup, and diagnosis and the results in the surgical pathology reports were used in this study. Complete absence of nuclear expression of MMR proteins (MLH1, MSH2, MSH6, and PMS2) in the cancerous nuclei in the context of appropriately reactive internal and external controls was considered as loss or deficiency.
Location of the background and/or adjacent gastric mucosa was determined by referring to endoscopic report and/or surgical pathology report. In some cases, biopsies taken within 6 months before the resection were included to assess the background gastric mucosa. The non-neoplastic gastric mucosa was evaluated using the updated Sydney system [9, 10]. The pathologic information collected included the presence and type of inflammation, presence of atrophy, presence of metaplasia (intestinal metaplasia (IM) for antral mucosa and body mucosa and pseudopyloric gland metaplasia for body mucosa), presence of enterochromaffin-like (ECL) cell hyperplasia, and H. pylori organisms. The histologic findings in the non-neoplastic gastric mucosa in this cohort of LS patients who developed gastric adenocarcinoma were compared to the previously published histologic findings in LS patients without gastric cancer [8]. The comparison was made using Fisher’s exact test, and a P value < 0.05 was considered statistically significant. Statistical analysis was performed using R2.15.2 (R Development Core Team, Vienna, Austria).
| Results | ▴Top |
Clinicodemographics of LS patients with gastric adenocarcinoma
As demonstrated in Table 1, our cohort of 11 LS patients included four men and seven women with gastric cancer. The mean age at the diagnosis of gastric adenocarcinoma was 63 years (range: 23 - 83). Diagnosis of LS was made clinically in two patients and the remaining nine patients were confirmed genetically (including MLH1 mutation (n = 3), MSH2 mutation (n = 3), MSH6 mutation (n = 1), PMS2 biallelic mutations (n = 1), MLH1 del/PMS2 mutation (n = 1)). Ten (90.9%) had a personal cancer history (colon cancer (n = 6), pancreatic cancer (n = 1), duodenal adenocarcinoma (n = 1), breast cancer (n = 2), endometrial cancer (n = 1), non-melanoma skin cancer (n = 2), medulloblastoma (n = 1), and cancer not further specified (n = 4)). Six (of seven, 85.7%) patients with available information had a family history of colon cancer. None of the nine patients with available information had a family history of gastric cancer.
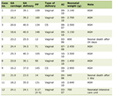 Click to view | Table 1. Clinicodemographics and Gastric Tumor Characteristics of LS Patients in This Study |
Six (54.5%) patients were symptomatic (abdominal pain (n = 2), epigastric discomfort (n = 1), reflux symptom (n = 1), dysphagia (n = 1), symptoms related to anemia (n = 1)) and six were in a surveillance program. One patient had a history of autoimmune diseases (one hyperthyroidism with positive rheumatoid factor). The remaining patients had no clinical evidence of autoimmune diseases. None of the patients had megaloblastic/pernicious anemia or were tested for serology for autoimmune gastritis, i.e., anti-parietal cell antibodies or anti-intrinsic factor antibodies. Seven patients were tested for H. pylori antibodies and all of them were negative, the other four patients were either not tested or had unknown results. None of the patients were tested for serum gastrin levels.
Tumor characteristics of gastric adenocarcinoma in LS patients
Eight (of 11, 72.7%) patients underwent gastrectomy and three received endoscopic resections. Tumor characteristics in the study subjects are summarized in Table 1. Nine (81.8%) patients had tumor in the fundus and/or body (fundus (n = 3), body (n = 5) and fundus and body (n = 1)). The other two patients had tumor in the antrum. Seven (of 11, 63.6%) cases were intestinal type or mixed and the remaining four were signet ring cell carcinoma/diffuse type. Adenocarcinoma invaded the mucosa in four cases, the submucosa in three, the muscularis propria in two, and the subserosa or at least subserosa in two. Eight gastrectomy cases had lymph nodes available for evaluation and none of them had nodal metastases. None of the patients had distant metastases at time of diagnosis.
Six of the cases were tested for MMR proteins (MLH1, MSH2, MSH6, and PMS2) via immunohistochemistry and all of them had at least one MMR protein loss (MLH1 (n = 3), MSH2 (n = 2), one with the lost MMR protein(s) (not specified).
Background gastric mucosa in LS patients
Data on background gastric mucosal changes are summarized in Table 2. Overall, eight (of 11, 72.7%) patients had chronic gastritis, five (45.4%) had atrophy, and four (36.3%) had IM. One case (of 11, 9.3%) had chronic active H. pylori gastritis. The gastric body was sampled in eight of 11 cases and five (of eight, 63%) had at least moderate chronic gastritis. Other findings in these gastric body biopsies included glandular atrophy (4/8, 50%), multifocal IM (3/8, 38%), and pseudopyloric gland metaplasia (4/8, 50%). ECL cell hyperplasia was noted in three of eight (37.5%) cases. The remaining three cases had histologically unremarkable body mucosa. Eight of the 11 cases had a gastric antrum biopsy available for review: two (of eight, 25%) had moderate chronic gastritis including one also with IM, two (of eight, 25%) mild chronic gastritis (25%), one (of eight, 13%) IM only, one (of eight, 13%) reactive gastropathy, and two (of eight, 25%) normal histology. Five of our cases had both antral and body biopsies performed. In these cases, four (80%) had a body predominant pattern of mucosal changes including three with IM; while one had normal antral and body mucosa. One case of a body-predominant chronic gastritis with associated dysplasia and MLH1- and PMS2-deficient adenocarcinoma in a MLH1-mutated LS patient is illustrated in Figure 1.
Click to view | Table 2. Background Gastric Mucosal Changes in LS Patients With Gastric Adenocarcinoma |
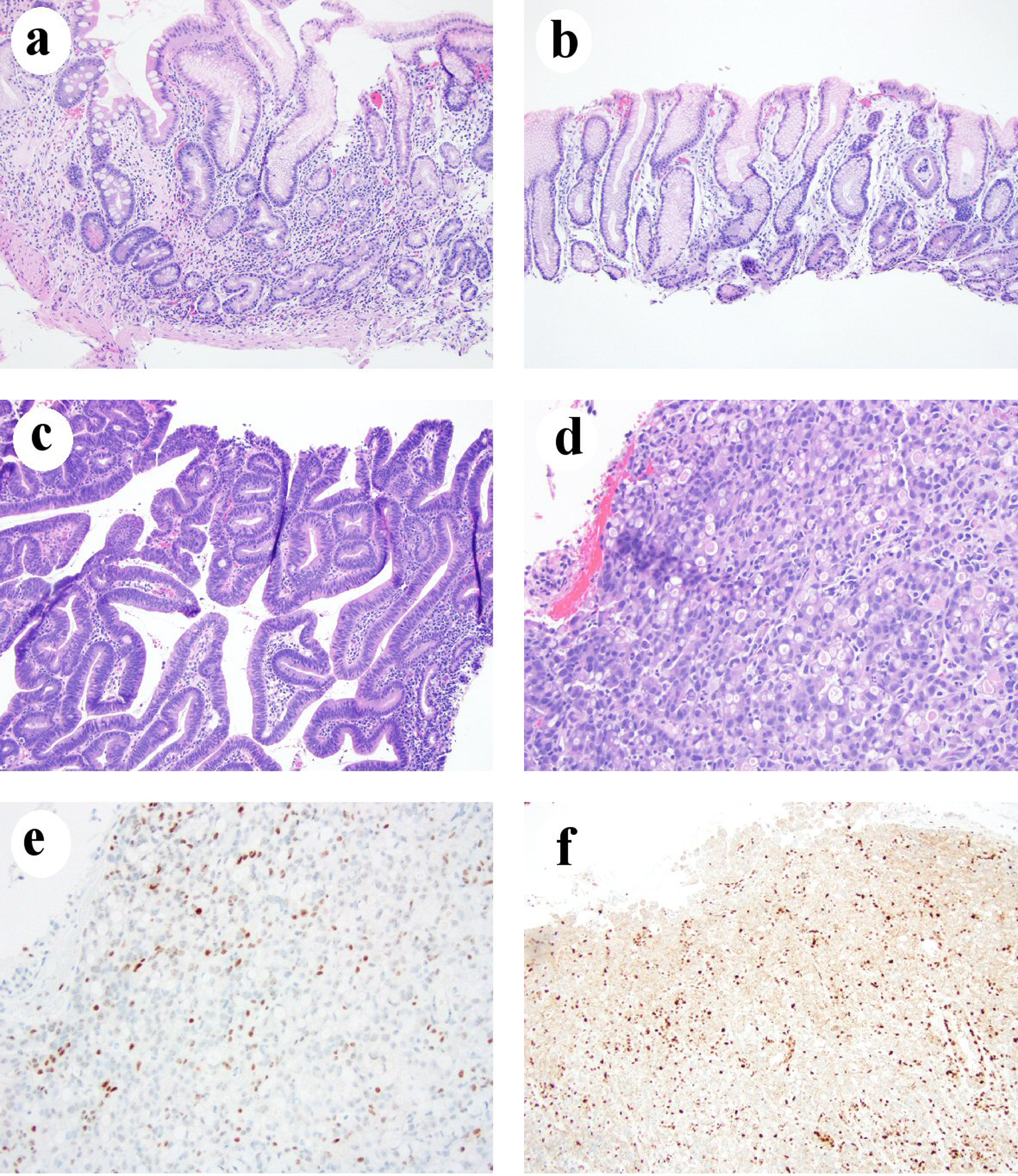 Click for large image | Figure 1. Chronic gastritis in one LS patient who developed gastric adenocarcinoma. (a) Body mucosa showing chronic inactive atrophic gastritis with intestinal metaplasia and pseudopyloric gland metaplasia (H&E stain, 100 ×). (b) Antral mucosa showing mild chronic inactive gastritis without intestinal metaplasia (H&E stain, 100 ×). (c) Histomorphology of low-grade dysplasia (H&E stain, 100 ×). (d) Signet ring cell/diffuse type carcinoma (H&E stain, 200 ×). (e) Loss of MLH1 in gastric adenocarcinoma nuclei (immunohistochemical stain, 200 ×). (f) Loss of PMS2 expression in gastric adenocarcinoma nuclei (immunohistochemical stain, 100 ×). H&E: hematoxylin and eosin; LS: Lynch syndrome. |
Compared to gastric mucosal abnormalities previously reported in LS patients without gastric cancer by Renkonen-Sinisalo et al (2002) [8], LS patients with gastric cancer in our current study tended to have a higher rate of chronic gastritis (P = 0.016), atrophy (P = 0.022), and a trend of a higher rate of IM, but this did not reach statistical significance (P = 0.08, Table 3 [8]).
Click to view | Table 3. Comparison of Gastric Mucosal Changes Between LS Patients With Gastric Cancer (Current Study) and Without Gastric Cancer From One Previous Study |
Corresponding to the inflammation being body predominant in this study, the majority of gastric tumors in our cohort arose in the gastric body or fundus (9/11 or 81.8%). Conversely, only two of our 11 cases had cancers arising in the gastric antrum. Infection with H. pylori was quite rare in our population, only one out of 11 (9.1%) patients had H. pylori infection at the time of biopsy/resection. Seven of our patients tested for H. pylori antibodies were negative.
Six (of 11, 54.5%) patients had precursor lesions (low-grade dysplasia (n = 3), high-grade dysplasia (n = 3)). Dysplasia arose from a background chronic gastritis in four patients, normal antral mucosa in one patient, and normal gastric body and fundic mucosa in one patient.
Gastritis in patients with gastric cancer and MLH1 mutations
Four patients in our cohort harbored germline MLH1 mutation (n = 3) or MLH1 deletion (n = 1), and all of them had chronic gastritis, with three of them being chronic autoimmune gastritis-like gastritis and one H. pylori gastritis. In the three cases with immune-mediated gastritis, all of them had atrophy, IM, and pseudopyloric gland metaplasia in the gastric body mucosa. In the case with H. pylori gastritis, the gastritis demonstrated a body predominant chronic active gastritis with IM but no pyloric gland metaplasia or ECL cell hyperplasia. It is not clear if that patient had been on a proton pump inhibitor.
Clinical follow-up
Three (of six, 50%) patients had GI symptom and two (of five, 40%) patients without GI symptoms had tumor invading muscularis propria or beyond (T2 or higher) (P =1). The follow-up period for these cases varied from 1 - 156 months (median 9 months). Two symptomatic patients and one patient in a surveillance program died at 1, 6, and 5 months after the diagnosis, respectively. The remaining patients were alive after a median follow-up of 12 months (range: 2 - 156 months).
| Discussion | ▴Top |
Hereditary nonpolyposis colorectal cancer (HNPCC or Lynch syndrome) is among the most common hereditary tumor syndromes, and the most common inherited GI cancer tumor syndrome [11]. One of the earliest descriptions of LS was from Aldred S Warthin in 1913, who described a family (family G) with a propensity of colorectal, uterine, and stomach cancers [11-13]. This “G family” was tracked over the years by several other physicians including Henry T Lynch, of whom the term “Lynch syndrome” was coined [12]. Breakthroughs in our molecular understanding of LS began in 1993, and by 1997 all four major MMR genes associated with LS were discovered [13]. As a result, currently LS is confirmed by germline mutational analysis of the MMR genes (MLH1, MSH2, MSH6, and PMS2) or other analysis of MMR genes when no deleterious mutations are identified by mutational analysis. In our series, nine cases were confirmed by germline MMR gene analysis, two cases were diagnosed based on strong personal and family cancer histories before the molecular testing were clinically available. We felt these two patients had LS-associated gastric adenocarcinomas as the tumors were MMR-deficient by immunohistochemistry (one with MSH2/MSH6 deficiency and one with the lost MMR protein(s) not specified). Nearly all (90.9%) patients in our cohort had a personal history of LS-associated malignancies (colonic, pancreatic, duodenal, breast, endometrial, non-melanoma cutaneous, medulloblastoma, etc.) and 85.7% had family history of colon cancer, indicating this is indeed a typical cohort of LS patients. In line with one previous study that LS patients with gastric cancer only rarely had a family history of gastric cancer [2], none of our nine LS patients who had available information in our cohort had a family history of gastric cancer.
Although gastric adenocarcinoma is a known LS-associated cancer, the exact gastric tumorigenesis remains unclear. Two major pathway theories exist for gastric adenocarcinoma: one through the chronic atrophic gastritis with intestinal metaplasia-dysplasia-adenocarcinoma sequence and the other due to mutation and resultant loss of E-cadherin function leading to diffuse type (or signet ring cell) gastric cancer [3]. Sporadic gastric adenocarcinoma is more often (about 70%) located in the antrum and lesser curvature or the lower portion of the stomach [14]. In our study, nine (of 11, 81.8%) adenocarcinomas were located in the fundus and/or body and 63.6% were intestinal or mixed type, consistent with previous reports [5, 7, 15]. The combination of intestinal type adenocarcinoma with a predilection for fundus/body suggests a tumorigenesis of body-predominant chronic gastritis-intestinal metaplasia, dysplasia, and carcinoma in LS patients, which is different from an antrum-predominant chronic H. pylori gastritis-intestinal metaplasia, dysplasia, and carcinoma in non-syndromic settings.
Corresponding to the majority of gastric tumors in our cohort which were located in the gastric body or fundus (9/11 or 81.8%), our study revealed body-predominant chronic gastritis in LS patients. More specifically, our study revealed 72.7% of LS patients with gastric adenocarcinoma had chronic gastritis and 45.4% with atrophy and 36.3% IM, consistent with one previous study that reported five of seven (71.4%) LS patients with gastric cancer had gastritis including three cases of autoimmune gastritis, one case of chronic atrophic gastritis but less prominent metaplasia and ECL hyperplasia, and one case of lymphocytic gastritis without evidence of H. pylori infection [7]. However, the topographical information of chronic atrophic gastritis in LS patients with gastric adenocarcinoma was not reported in that report [7]. Interestingly, all of the LS patients with chronic gastritis in that report harbored mutations in MSH2 which have been reported to be associated with higher risk of developing gastric cancer in LS patients [2]. In our study, four patients had germline MLH1 alterations (mutation or deletion); three of them had chronic immune-mediated gastritis and one H. pylori gastritis. Our results were similar to one previous study that 93% LS patients with gastric adenocarcinoma had gastric atrophy and 86% with IM [6]. Taken together, these results support IM-dysplasia-carcinoma as the tumorigenesis in the majority of gastric cancer in LS patients with MLH1 or MSH2 mutations. More importantly, most chronic gastritis in LS patients with gastric cancer are immune-mediated as four out of five cases in our cases with body and antrum biopsies showed a pattern of body predominant gastritis, similar to autoimmune gastritis although neither serologic testing for autoimmune gastritis nor gastrin levels was performed in any cases in our study. The etiology for chronic immune-mediated gastritis in LS patients remains unknown; however, an association of LS with autoimmune diseases has been suggested in previously studies [16, 17]. Indeed, one of our patients had hyperthyroidism, and positive serum rheumatoid factor, and chronic atrophic gastritis with pseudopyloric gland metaplasia and ECL hyperplasia but without IM in the body. Serology for H. pylori was negative, but the antrum was not sampled. The body-predominant gastritis in LS patients with gastric adenocarcinoma in our study is different from an earlier study which revealed the presence of antral atrophic chronic gastritis with IM [4]. The discrepancy may be due to geographic difference (Italy vs. USA in this study) and the era of diagnosis (years prior to mid 1980s when H. pylori infection was not recognized as the etiology of antrum-predominant chronic gastritis).
Our study also revealed one case (of 11, 9.1%) of H. pylori gastritis in LS patients with gastric adenocarcinoma. This rate is similar to previously reported H. pylori gastritis rate (10-27%) in LS patients [15, 18], LS patients with gastric cancer [5, 6], LS patients without gastric cancer [7] or mutation carriers, and mutation negative family members of LS patients [8]. These results suggest that MMR gene mutations are not associated with increased risk of H. pylori infection. Interesting, the only H. pylori gastritis case in our study had fundus-located adenocarcinoma and a body-predominant chronic active gastritis, a pattern different from classic antrum-predominant H. pylori gastritis. Possibilities for this pattern include long-term H. pylori infection, H. pylori gastritis with superimposed autoimmune gastritis and long-term use of proton pump inhibitor shifting the chronic gastritis to the body. No serologic studies for autoimmune gastritis were performed in this patient. Similar to this H. pylori gastritis case in our series, three of four previously reported LS patients with H. pylori infection had tumor located in the corpus in two (66.7%) cases suggesting an extension of chronic H. pylori gastritis into the corpus [15] leading to the cancer development in the corpus. These results suggest that LS patients with H. pylori gastritis should be treated with anti-H. pylori therapy to eradicate the infection and to reduce the risk of gastric cancer.
Our study provides additional evidence for a chronic immune-mediated gastritis in LS patients who developed gastric adenocarcinoma, substantiating the findings in one previous study consisted primarily of LS patients with MSH2 mutation [7]. Increased risk for gastric adenocarcinoma is observed in LS patients but the incidence rate is still low [2], which challenges the universal use of gastric cancer surveillance program in LS patients as it may not be cost-effective [18]. Our data and previous data suggest that LS patients may be further risk stratified for gastric cancer based on the specific MMR gene mutated (mutations in MLH1 and MSH2 carry higher risk), H. pylori infection, the presence of immune-mediated gastritis or gastric IM, and family history of gastric cancer. Reviewing the literature only identified one LS patient with gastric cancer and chronic gastritis was tested and positive for anti-intrinsic factor [7]. Further prospective studies incorporating serologic testing (autoimmune gastritis and H. pylori), gastrin levels, vitamin 12 levels, and systemic biopsy (fundus, body, and antrum) and interpretation of gastric biopsy using the updated Sydney system are needed to help optimize and individualize the gastric cancer surveillance program for LS patients.
Autoimmune gastritis with atrophy and IM has an increased risk for gastric adenocarcinoma, but the etiology of autoimmune gastritis and autoimmune gastritis-associated tumorigenesis remain unclear. One recent study showed about 21% of autoimmune gastritis-associated gastric adenocarcinomas were MMR deficient, but germline mutational analysis was not performed on those patients [19]. The high frequency of MMR-deficient gastric adenocarcinoma in patients with autoimmune gastritis and our finding of gastric adenocarcinoma arising from an immune-mediated atrophic gastritis in LS patients suggest that germline mutation of MMR genes may be an etiology of autoimmune gastritis with a predilection for developing MMR-deficient gastric adenocarcinoma, at least in some cases. This hypothesis needs additional studies.
Our study has several strengths. First, all cases were diagnosed by clinical personal and family cancer history with most cases further being confirmed genetically. In addition, pathologic report and slides were reviewed by GI pathologists who had GI pathology fellowship training and have been practicing in hospitals with large volume of gastric specimens. Further, most cases were diagnosed in the past 10 years from 2010 - 2019 which allows confident diagnosis of H. pylori infection and confirmation of MMR deficiency in the tumor by immunohistochemistry.
Our study has several limitations. First, it only had 11 LS patients with gastric cancer. This is partly due to the rarity of the disease and our strict inclusion criteria. Second, patients in this cohort were from five institutions, and this may have resulted in non-uniform information on medication, personal and family cancer history and laboratory testing for autoimmune gastritis and H. pylori serology. There may have been referral bias as these are large referral medical centers. Third, the pathology material was only reviewed by one GI pathologist at the contributing institution without further central review.
In summary, our study revealed that none of LS patients with gastric adenocarcinoma had a family history of gastric cancer. Most gastric adenocarcinoma in LS patients were located in the fundus and/or body and most of them are intestinal type with a tumorigenesis of chronic gastritis (immune gastritis in most cases and H. pylori gastritis in the minority of cases)-associated IM, dysplasia, and carcinoma. LS-associated autoimmune gastritis needs to be further characterized both clinically and by laboratory testing in future studies, in addition to testing for H. pylori infection and using the biopsy protocol recommended in the updated Sydney system [20]. Prospective studies are urgently needed to address the use of gastric cancer surveillance program in LS patients as an increasing number of LS patients are diagnosed through universal testing and screening newly diagnosed colorectal cancer for LS.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
None to declare.
Informed Consent
Not applicable.
Author Contributions
DS: data collection, analysis and manuscript drafting; RC, KW, Xuefeng Zhang, MW, JC, JL, Xuchen Zhang, MF and MS: data collection and critical review of manuscript; XL: study design and supervision, data analysis and manuscript drafting.
Data Availability
The authors declare that data supporting the findings of this study are available within the article.
| References | ▴Top |
- Gylling A, Abdel-Rahman WM, Juhola M, Nuorva K, Hautala E, Jarvinen HJ, Mecklin JP, et al. Is gastric cancer part of the tumour spectrum of hereditary non-polyposis colorectal cancer? A molecular genetic study. Gut. 2007;56(7):926-933.
doi pubmed - Capelle LG, Van Grieken NC, Lingsma HF, Steyerberg EW, Klokman WJ, Bruno MJ, Vasen HF, et al. Risk and epidemiological time trends of gastric cancer in Lynch syndrome carriers in the Netherlands. Gastroenterology. 2010;138(2):487-492.
doi pubmed - Sipponen P, Hyvarinen H, Seppala K, Blaser MJ. Review article: Pathogenesis of the transformation from gastritis to malignancy. Aliment Pharmacol Ther. 1998;138(Suppl 1):61-71.
doi pubmed - Cristofaro G, Lynch HT, Caruso ML, Attolini A, DiMatteo G, Giorgio P, Senatore S, et al. New phenotypic aspects in a family with Lynch syndrome II. Cancer. 1987;60(1):51-58.
doi - Aarnio M, Salovaara R, Aaltonen LA, Mecklin JP, Jarvinen HJ. Features of gastric cancer in hereditary non-polyposis colorectal cancer syndrome. Int J Cancer. 1997;74(5):551-555.
doi - Lee SE, Kang SY, Cho J, Lee B, Chang DK, Woo H, Kim JW, et al. Pyloric gland adenoma in Lynch syndrome. Am J Surg Pathol. 2014;38(6):784-792.
doi pubmed - Adar T, Friedman M, Rodgers LH, Shannon KM, Zukerberg LR, Chung DC. Gastric cancer in Lynch syndrome is associated with underlying immune gastritis. J Med Genet. 2019;56(12):844-845.
doi pubmed - Renkonen-Sinisalo L, Sipponen P, Aarnio M, Julkunen R, Aaltonen LA, Sarna S, Jarvinen HJ, et al. No support for endoscopic surveillance for gastric cancer in hereditary non-polyposis colorectal cancer. Scand J Gastroenterol. 2002;37(5):574-577.
doi pubmed - Sipponen P, Price AB. The Sydney System for classification of gastritis 20 years ago. J Gastroenterol Hepatol. 2011;26(Suppl 1):31-34.
doi pubmed - Rugge M, Pennelli G, Pilozzi E, Fassan M, Ingravallo G, Russo VM, Di Mario F, et al. Gastritis: the histology report. Dig Liver Dis. 2011;43(Suppl 4):S373-384.
doi - Boland CR, Yurgelun MB. Historical perspective on familial gastric cancer. Cell Mol Gastroenterol Hepatol. 2017;3(2):192-200.
doi pubmed - Douglas JA, Gruber SB, Meister KA, Bonner J, Watson P, Krush AJ, Lynch HT. History and molecular genetics of Lynch syndrome in family G: a century later. JAMA. 2005;294(17):2195-2202.
doi pubmed - Lynch PM. History of hereditary nonpolyposis colorectal cancer or "Lynch Syndrome". Revista Medica Clinica Las Condes. 2017;28(4):500-511.
doi - Kim SJ, Choi CW. Common Locations of Gastric Cancer: Review of Research from the Endoscopic Submucosal Dissection Era. J Korean Med Sci. 2019;34(35):e231.
doi pubmed - Fornasarig M, Magris R, De Re V, Bidoli E, Canzonieri V, Maiero S, Viel A, et al. Molecular and Pathological Features of Gastric Cancer in Lynch Syndrome and Familial Adenomatous Polyposis. Int J Mol Sci. 2018;19(6):1682.
doi pubmed - Rahner N, Hoefler G, Hogenauer C, Lackner C, Steinke V, Sengteller M, Friedl W, et al. Compound heterozygosity for two MSH6 mutations in a patient with early onset colorectal cancer, vitiligo and systemic lupus erythematosus. Am J Med Genet A. 2008;146A(10):1314-1319.
doi pubmed - Caruso ML, Cristofaro G, Lynch HT. HNPCC-Lynch syndrome and idiopathic inflammatory bowel disease. A hypothesis on sharing of genes. Anticancer Res. 1997;17(4A):2647-2649.
- Galiatsatos P, Labos C, Jeanjean M, Miller K, Foulkes WD. Low yield of gastroscopy in patients with Lynch syndrome. Turk J Gastroenterol. 2017;28(6):434-438.
doi pubmed - Henn P, Ma C. Gastric carcinomas arising in autoimmune metaplastic atrophic gastritis are associated with increased frequency of DNA mismatch repair deficiency and EBV infection. Abstracts from USCAP 2019: Gastrointestinal Pathology (578-775). Mod Pathol. 2019;32(supplement 2):1-164, page 57.
doi pubmed - Dixon MF, Genta RM, Yardley JH, Correa P. Classification and grading of gastritis. The updated Sydney System. International Workshop on the Histopathology of Gastritis, Houston 1994. Am J Surg Pathol. 1996;20(10):1161-1181.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Gastroenterology Research is published by Elmer Press Inc.