

| Gastroenterology Research, ISSN 1918-2805 print, 1918-2813 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Gastroenterol Res and Elmer Press Inc |
| Journal website https://www.gastrores.org |
Case Report
Volume 17, Number 4, August 2024, pages 189-193

Adult-Onset Autoimmune Enteropathy Mimicking Disaccharidase Deficiency
Georgeta Giblena, Jerry Huangb, Brandon Yuc, Jinping Laid, e
aMercy Medical Center, Baltimore, MD 21002, USA
bGilman School, Baltimore, MD 21210, USA
cRiver Hill High School, Clarksville, MD 21029, USA
dDepartment of Pathology, Kaiser Permanente Sacramento Medical Center, CA 95825, USA
eCorresponding Author: Jinping Lai, Department of Pathology and Laboratory Medicine, Kaiser Permanente Sacramento Medical Center, Sacramento, CA 95825, USA
Manuscript submitted May 1, 2024, accepted July 1, 2024, published online July 18, 2024
Short title: Adult Autoimmune Enteropathy
doi: https://doi.org/10.14740/gr1737
| Abstract | ▴Top |
Adult-onset autoimmune enteropathy is a rare disorder characterized by severe diarrhea, weight loss, malnutrition, and enteric mucosal atrophy resulting from immune-mediated injury. Disaccharidase deficiencies are a group of disorders characterized by inadequate activity of disaccharidase in the small intestine, leading to impaired digestion of disaccharides and malabsorption. Here, we present a case of adult-onset autoimmune enteropathy initially diagnosed as disaccharidase deficiency based on the clinical symptoms of chronic diarrhea, weight loss, and severely reduced levels of lactase, maltase, palatinase, and sucrase in duodenal aspirates. However, follow-up duodenal biopsy revealed markedly villous blunting, goblet and Paneth cell depletion, increased crypt apoptotic bodies and lamina propria lymphoplasmacytic inflammation, leading to a revised diagnosis of autoimmune enteropathy. This case highlights the diagnostic challenges of adult-onset autoimmune enteropathy and the importance of considering it in adults with unexplained gastrointestinal symptoms. It also emphasizes the need for tissue biopsies in cases with inconclusive initial diagnostic tests. Increased awareness of these disorders and their mimickers can improve diagnosis and management, ultimately benefiting patients with these conditions.
Keywords: Autoimmune enteropathy; Disaccharidase deficiency; Malnutrition
| Introduction | ▴Top |
Adult-onset autoimmune enteropathy (AIE) is a rare cause of chronic diarrhea, weight loss, and malnutrition secondary to immune-mediated damage to the intestinal mucosa [1]. In AIE, abnormal expression of self- antigens on enterocyte activates CD4 T lymphocytes, leading to the destruction of the enterocytes through apoptosis or other cytotoxic effect. This results in impaired nutrient absorption and severe gastrointestinal symptoms [2, 3]. Most patients (up to 79%) with AIE often present with concomitant immunodeficiency disorder and other autoimmune-related diseases [1-3]. Refined diagnostic criteria for AIE include severe diarrhea, villous atrophy, lack of response to dietary restrictions (especially gluten), and a predisposition to autoimmunity, while excluding medication/drug-induced secondary AIE and other immunodeficiency disorders [1, 4]. Literatures suggest that anti-enterocyte antibodies may be no longer considered necessary for the diagnosis of AIE [3, 5]. Endoscopic findings can have a normal or nonspecific atrophic appearance. The histological patterns of AIE can be heterogeneous, and the characteristic histological findings may include villous blunting, increased lamina propria lymphoplasmacytic inflammation, crypt epithelial apoptosis, and intraepithelial lymphocytosis [6]. Diagnosis of AIE remains challenging due to significant overlap with other diseases and lack of specific clinical, endoscopic, and histopathologic findings.
Disaccharidase deficiencies are a group of disorders characterized by reduced activity of disaccharidase enzymes in the small intestine [7]. These enzymes are mainly located at the duodenal brush border and are responsible for breaking down specific disaccharides into simpler sugars that can be absorbed. Insufficient enzymes can lead to impaired digestion of disaccharides such as lactose, sucrose, maltose, and galactose, resulting in abdominal pain, bloating, diarrhea, and malnutrition [8]. Although more recognized in infants and children, these deficiencies can also occur in adults. The diagnosis of disaccharidase deficiency may include disaccharidase assay, hydrogen breath testing, and disaccharidase challenge. The diagnosis should be considered in patients with post-prandial abdominal pain, gas, bloating, and diarrhea. Secondary causes of disaccharidase deficiency, such as AIE, celiac, and Crohn’s disease, acute gastroenteritis, and other conditions which may damage the brush border should be excluded. Additionally, genetic testing to exclude congenital deficiency should be considered, especially in the pediatric population [9]. Congenital sucrase-isomaltase deficiency is a rare genetic form of disaccharide malabsorption. Interestingly, some particular sucrase-isomaltase (SI) gene variants can lead to reduced enzymatic activity and predispose to irritable bowel syndrome [10].
This paper presents a case of adult-onset AIE initially diagnosed as disaccharidase deficiency, highlighting diagnostic challenges and emphasizing the importance of a systematic approach to evaluation, aiming to improve the diagnosis and management of AIE in adults.
| Case Report | ▴Top |
A 27-year-old Caucasian man who has a complex medical history of scleroderma (pansclerotic morphea) with poor response to immunosuppressive monotherapy (rituximab, Otezla or tocilizumab). He also had a history of pancreatic insufficiency, failure to thrive in adulthood, hypothyroidism, attention-deficit hyperactivity disorder (ADHD), and dermatomyositis. In late 2023, he had a prolonged hospitalization at John Hopkins University Hospital for several issues, most notably chronic diarrhea and pneumatosis intestinalis. Prior to the hospitalization, an extensive gastrointestinal workup including serologies for celiac disease and infection, was conducted and revealed negative results. A trial gluten-free diet failed to alleviate his gastroenterological symptoms. During the hospitalization, he developed a small bowel obstruction and fungal infection. Subsequently, he presented himself in the Gastrointestinal Motility Clinic at Mercy Medical Center with difficulty in swallowing, intermittent severe abdominal pains, chronic diarrhea, bloating, and a significant weight loss of 35 pounds over 1 year. His physical examination showed a thin, malnourished young patient in his wheelchair. His lab workups showed low hemoglobin (Hb) (9.2 g/dL, normal: 13.9 - 16.3 g/dL), low total protein (4.4 g/dL, normal: 6.0 - 8.0 g/dL), low albumin (2.4 g/dL, normal: 3.5 - 5.3 g/dL), and a decreased vitamin A (20 µg/dL, normal: 38 - 98 µg/dL). His total immunoglobulin G (IgG), IgA, and IgM are normal. The tests for infections including human immunodeficiency virus (HIV), varicella zoster virus, Giardia, and Clostridium difficile were negative.
An upper gastrointestinal (GI) endoscopy was performed and showed an esophageal web, unremarkable stomach, and slightly atrophic duodenum (Fig. 1). However, the duodenal aspirate showed severely reduced enzymes with lactase 0.58 (normal > 14.0), maltase 44.16 (normal > 110), palatinase 2.63 (normal > 8.5), and sucrase 2.05 (normal > 25), suggesting disaccharidase deficiencies. Meanwhile, the duodenal aspirate cultures showed bacterial overgrowth with > 100,000 colony-forming units (CFU)/mL of diphtheroids, Streptococcus viridans group, Haemophilus species, Lactobacillus species, and mixed anaerobic flora. A diagnosis of disaccharidase deficiencies with a component of small bowel bacterial overgrowth was initially made.
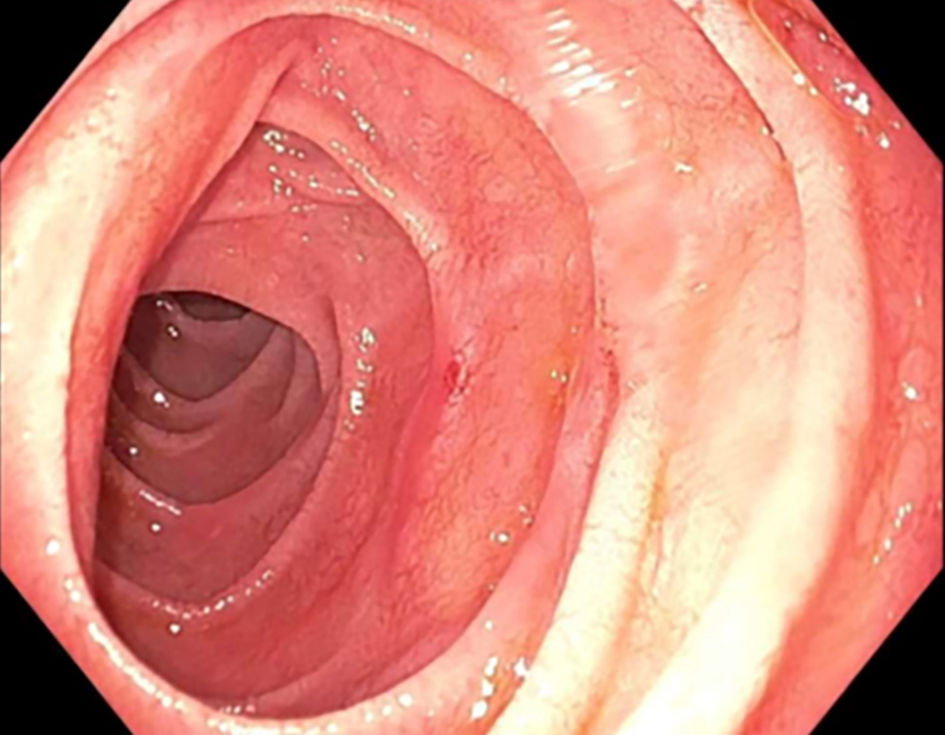 Click for large image | Figure 1. Upper gastrointestinal endoscopy showing slightly atrophic duodenum. |
Subsequent duodenal biopsies showed moderate to severe villous blunting, increased lamina propria lymphoplasmacytic inflammation, crypt hyperplasia, complete loss of goblet cells and Paneth cells (Fig. 2a, b), prominent crypt apoptosis (Fig. 2c), and mild intraepithelial lymphocytosis (Fig. 2d). These histologic findings, along with the patient’s clinical history of systemic scleroderma, hypothyroidism, and pancreatic insufficiency, were most likely consistent with adult-onset AIE. The anti-enterocyte antibody and anti-goblet cell antibody were not tested due to the unavailability in our hospital lab and our contracted reference lab. The patient started on budesonide 9 mg/day and rifaximin 550 mg twice daily for 4 weeks followed by gradually tapering budesonide 6 mg/day for additional 6 weeks, the patient revealed significant clinical improvement including decreased abdominal pain, diarrhea, and a stable weight.
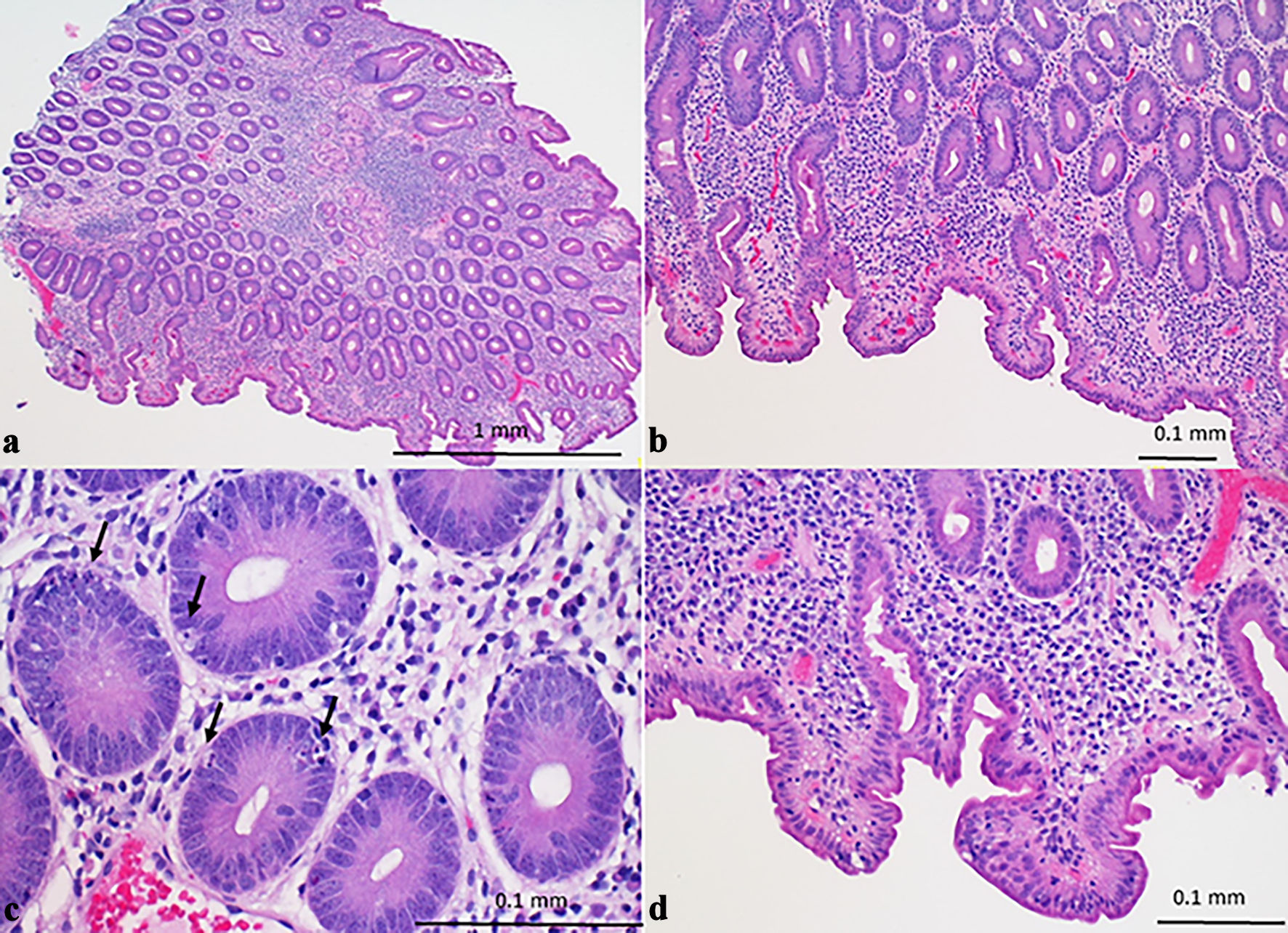 Click for large image | Figure 2. Duodenal biopsy (hematoxylin and eosin stain) showing chronic active enteropathy with moderate to severe villous blunting, lamina propria lymphoplasmacytic inflammation, neutrophilic cryptitis (a), goblet cell and Paneth cell depletion (b), increased crypt epithelial apoptosis (arrows) (c), and intraepithelial lymphocytosis (d). |
| Discussion | ▴Top |
AIE is a rare immune-mediated disorder characterized by inflammation of the duodenal mucosa causing intractable diarrhea, bloating, abdominal pain, and malnutrition [1]. It is primarily a pediatric disease, but now, it is well recognized to involve adults [2]. The adult-onset AIE appears to affect adults of all ages, with a slight female predominance reported in some studies. The patients are usually not responsive to dietary modification (mainly a gluten-free diet). In addition, AIE was frequently encountered in the setting of immunodeficiency disorders, and patients may also present with extra-intestinal manifestations such as autoimmune thyroiditis or dermatitis herpetiformis [3, 4].
The diagnosis of adult-onset AIE can be challenging and often requires a combination of clinical, serological, endoscopic, and histological findings. Current diagnostic criteria may include chronic diarrhea (> 6 weeks duration), malabsorption, specific duodenal histopathologic findings, and exclusion of other causes of villous atrophy [1].
The endoscopic findings can be totally normal or present some nonspecific changes including mucosal erythema, edema, erosions, and atrophy. Histological examination of duodenal biopsies is the gold standard diagnostic test, with characteristic features of villous blunting, crypt hyperplasia, lymphocytic infiltration of the lamina propria, and goblet cell and Paneth cell depletion [6]. However, the histologic findings are largely diverse and can be further classified into four categories with descending prevalence rates: active chronic enteritis (52%), celiac disease-like (20%), graft-versus-host disease-like (16%), and mixed/no predominant pattern (12%). Additionally, 64% of patients can present with colonic inflammation [6]. In order to make a definite diagnosis of AIE, it is necessary to exclude other causes of villous atrophy, e.g., celiac disease, refractory celiac disease, seronegative celiac disease, and medication (olmesartan)-associated enteropathy [6].
Distinguishing between celiac disease and AIE relies on their distinct clinical, pathological, and immunological features. Clinically, celiac disease typically presents with chronic diarrhea, weight loss, abdominal pain, and sometimes iron deficiency anemia [11]. Whereas AIE is often seen in younger patients and presents with severe, chronic diarrhea, and malnutrition. AIE may occur with other autoimmune disorders such as IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X-linked) syndrome [2, 3]. Pathologically, celiac disease is characterized by villous atrophy, crypt hyperplasia, and increased intraepithelial lymphocytes in the small intestine mucosa. While AIE also shows villous atrophy but with more pronounced crypt apoptosis and a high level of lymphocyte infiltration in the lamina propria [11]. Additionally, celiac disease can be diagnosed by the presence of specific antibodies such as anti-tissue transglutaminase (tTG) and anti-endomysial antibodies (EMA) [12]. The genetic markers human leukocyte antigen (HLA)-DQ2 and HLA-DQ8 are also associated with celiac disease [13]. Celiac disease can be managed with a strict gluten-free diet, which typically leads to clinical and histological improvement. AIE usually is not responsive to gluten-free diet and requires immunosuppressive therapy, such as corticosteroids, tacrolimus, or cyclosporine. In summary, while both conditions share some similarities, they can be distinguished based on clinical presentation, pathological features, specific immunological markers, and their response to treatment.
The presence of anti-enterocyte or anti-goblet cell antibodies increases the likelihood of AIE but is not required to establish the diagnosis, as these antibodies are not consistently present. In pediatric patients with AIE, 79% had positive anti-enterocyte antibodies, while adult patients with AIE showed that 27% with anti-goblet cell antibodies and 60% with anti-enterocyte antibodies [3, 5]. Meanwhile, since there is no correlation between disease severity by histology and autoantibody titers, the clinical utility of autoantibodies as a marker for disease severity or treatment effect is limited. Additionally, the autoantibodies may only appear at certain times during the disease course and disappear prior to the resolution of the enteropathy. Autoantibodies may also disappear with the initiation of therapy, even if mucosal healing has not occurred [5]. Overall, from a practical perspective, the presence or absence of autoantibodies is not required for diagnosis of AIE but may help to narrow down the diagnosis.
Interestingly, our patient’s AIE was misdiagnosed with disaccharidase deficiency. To the best of our knowledge, this is the first case of AIE being diagnosed with disaccharidase deficiency due to its significant overlapping clinical symptoms.
Disaccharidases are a group of enzymes responsible for the glycosidic hydrolysis of disaccharides (lactose, sucrose, maltose, etc.) into monosaccharides (glucose, galactose, and fructose). The monosaccharides are transported across the epithelial brush border to be absorbed and metabolized. Disaccharidase deficiency can cause increased osmotic load from mal-absorbed disaccharides in the small intestine, and lead to diarrhea and abdominal pain. The undigested disaccharides are further fermented by colonic bacteria, causing the production of gas and bloating.
The main reason that our patient’s AIE was initially misdiagnosed as disaccharidase deficiency is due to the presence of villous atrophy. Since disaccharidases are predominantly found in the intestinal brush border, the lack of villi and brush border will cause a secondary deficiency in disaccharidases and resultant reduction of disaccharidase level in the disaccharidase assay. The undigested disaccharides further cause disaccharidase deficiency-like symptoms. The presence of goblet cell and Paneth cell depletion, along with other histologic features can distinguish AIE from disaccharidase deficiency in the duodenal biopsies, as in this case.
The exact pathogenesis of AIE remains unclear, but it is generally believed to involve an abnormal immune response targeting the enteric mucosa, especially mal-regulation of T cell-mediated immune system [3]. It is often associated with IPEX syndrome [3, 4]. A few other autoantibodies, including antinuclear, microsomal, and anti-smooth muscle antibodies, were reported positive in AIE patients [5].
The management of adult-onset AIE is primarily aimed at controlling symptoms and reducing inflammation [1]. This typically involves immunosuppressive therapy with corticosteroids or other immunomodulatory agents. Typically, steroids for 8 weeks of therapy are effective in up to 60% of AIE patients. Other common immunosuppressive medicines may include infliximab, 6-mercaptopurine, azathioprine, tacrolimus, and cyclosporine [3]. Dietary modifications, such as avoiding gluten, may also be beneficial in some patients. In severe cases, surgical intervention may be required to manage complications such as strictures or perforation. A timely diagnosis is important to start appropriate treatment and avoid more severe complications.
In summary, adult-onset AIE is a rare autoimmune disorder that can present with a wide range of symptoms and complications. Early recognition and diagnosis are crucial for initiating appropriate treatment and improving patient outcomes. Further research is needed to better understand the pathogenesis of this condition and to develop more effective treatment strategies.
Acknowledgments
None to declare.
Financial Disclosure
There is no financial disclosure regarding this case report.
Conflict of Interest
The authors declare no conflict of interest.
Informed Consent
Informed consent was obtained from the patient.
Author Contributions
G. Giblen drafted the manuscript and reviewed the literature. J. Huang collected and analyzed the data, reviewed the literature, and drafted the article. B. Yu collected and analyzed the data. J. Lai designed the study and finalized the article.
Data Availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request.
| References | ▴Top |
- Akram S, Murray JA, Pardi DS, Alexander GL, Schaffner JA, Russo PA, Abraham SC. Adult autoimmune enteropathy: Mayo Clinic Rochester experience. Clin Gastroenterol Hepatol. 2007;5(11):1282-1290.
doi pubmed pmc - Gentile NM, Murray JA, Pardi DS. Autoimmune enteropathy: a review and update of clinical management. Curr Gastroenterol Rep. 2012;14(5):380-385.
doi pubmed pmc - Chen CB, Tahboub F, Plesec T, Kay M, Radhakrishnan K. A review of autoimmune enteropathy and its associated syndromes. Dig Dis Sci. 2020;65(11):3079-3090.
doi pubmed - van Wanrooij RLJ, Neefjes-Borst EA, Bontkes HJ, Schreurs MWJ, Langerak AW, Mulder CJJ, Bouma G. Adult-onset autoimmune enteropathy in an European tertiary referral center. Clin Transl Gastroenterol. 2021;12(8):e00387.
doi pubmed pmc - Montalto M, D'Onofrio F, Santoro L, Gallo A, Gasbarrini A, Gasbarrini G. Autoimmune enteropathy in children and adults. Scand J Gastroenterol. 2009;44(9):1029-1036.
doi pubmed - Masia R, Peyton S, Lauwers GY, Brown I. Gastrointestinal biopsy findings of autoimmune enteropathy: a review of 25 cases. Am J Surg Pathol. 2014;38(10):1319-1329.
doi pubmed pmc - Viswanathan L, Rao SSC, Kennedy K, Sharma A, Yan Y, Jimenez E. Prevalence of disaccharidase deficiency in adults with unexplained gastrointestinal symptoms. J Neurogastroenterol Motil. 2020;26(3):384-390.
doi pubmed pmc - El-Chammas K, Williams SE, Miranda A. Disaccharidase deficiencies in children with chronic abdominal pain. JPEN J Parenter Enteral Nutr. 2017;41(3):463-469.
doi pubmed - Gericke B, Amiri M, Scott CR, Naim HY. Molecular pathogenicity of novel sucrase-isomaltase mutations found in congenital sucrase-isomaltase deficiency patients. Biochim Biophys Acta Mol Basis Dis. 2017;1863(3):817-826.
doi pubmed - Henstrom M, Diekmann L, Bonfiglio F, Hadizadeh F, Kuech EM, von Kockritz-Blickwede M, Thingholm LB, et al. Functional variants in the sucrase-isomaltase gene associate with increased risk of irritable bowel syndrome. Gut. 2018;67(2):263-270.
doi pubmed pmc - Sharma A, Choung RS, Wang XJ, Russo PA, Wu TT, Nehra V, Murray JA. Features of adult autoimmune enteropathy compared with refractory celiac disease. Clin Gastroenterol Hepatol. 2018;16(6):877-883.e871.
doi pubmed - Volta U, Mumolo MG, Caio G, Boschetti E, Latorre R, Giancola F, Paterini P, et al. Autoimmune enteropathy: not all flat mucosa mean coeliac disease. Gastroenterol Hepatol Bed Bench. 2016;9(2):140-145.
pubmed pmc - Sollid LM, Lie BA. Celiac disease genetics: current concepts and practical applications. Clin Gastroenterol Hepatol. 2005;3(9):843-851.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Gastroenterology Research is published by Elmer Press Inc.