

| Gastroenterology Research, ISSN 1918-2805 print, 1918-2813 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Gastroenterol Res and Elmer Press Inc |
| Journal website https://www.gastrores.org |
Case Report
Volume 15, Number 1, February 2022, pages 33-38

Polygenic Infantile Juvenile Polyposis Syndrome Managed With Sirolimus and Endoscopic Polypectomy
Lisa Dillon Bella, John A. Bernatb, Riad Rahhalc, d
aUniversity of Iowa Carver College of Medicine, Iowa City, IA 52242, USA
bDivision of Medical Genetics and Genomics, University of Iowa Stead Family Children’s Hospital, Iowa City, IA 52242, USA
cDivision of Pediatric Gastroenterology, University of Iowa Stead Family Children’s Hospital, Iowa City, IA 52242, USA
dCorresponding Author: Riad Rahhal, Division of Pediatric Gastroenterology, University of Iowa Stead Family Children’s Hospital, Iowa City, IA 52242, USA
Manuscript submitted November 11, 2021, accepted December 23, 2021, published online January 10, 2022
Short title: Polygenic Infantile JPS Managed With Sirolimus
doi: https://doi.org/10.14740/gr1480
| Abstract | ▴Top |
In the following clinical case of infantile juvenile polyposis syndrome (JPS), administration of a pharmacologic agent sirolimus was associated with reduced disease burden without need for bowel resection. The positive impact included improvement in protein-losing enteropathy, decreased intestinal blood loss, and improved weight gain. In addition, the number of polyps resected per unit time and frequency of upper and lower endoscopic evaluation needed dropped after initiation of sirolimus. This case report describes a positive clinical outcome and discusses the use of sirolimus with aggressive polypectomy as a potential treatment for the rare disease entity of polygenic infantile JPS. Through this case, we aim to emphasize that while administration of this drug may mitigate many sequelae of infantile JPS, it does not appear to eliminate the need for aggressive polypectomy.
Keywords: JPS; PTEN; BMPR1A; Hamartoma; PLE; mTOR; Rapamycin
| Introduction | ▴Top |
Juvenile polyposis syndrome (JPS) is a rare autosomal dominant disease associated with hamartomatous growths that can occur throughout the gastrointestinal (GI) tract of affected children [1, 2]. Infantile JPS is a more aggressive form that arises during the first year of life, and is thought to be caused by deletion of the PTEN and/or BMPR1A genes [3-5]. Without intervention, infantile JPS is often associated with detrimental health sequelae such as protein losing enteropathy (PLE), blood loss, malabsorption, malnutrition, and intussusception. Extensive medical and surgical interventions are needed to mitigate the sequelae of the disease, including parenteral nutrition (PN) support, frequent transfusions of red blood cells and albumin, endoscopic polypectomies, and bowel resection [4-6].
Previous case reports have demonstrated some success in treating juvenile polyposis with multi-organ transplantation, colectomy with endoscopic polypectomy, and even sirolimus with subtotal colectomy and sirolimus alone [4-7]. Our case report characterizes the complete clinical course of one patient with this rare disease entity. In the following clinical case, administration of a pharmacologic agent, sirolimus, successfully reduced GI polyp burden allowing for weaning off PN support and avoiding the need for bowel resection.
This case demonstrates a positive clinical outcome with early use of sirolimus along with aggressive polypectomy to manage infantile JPS. It serves to both highlight the benefit of using sirolimus, in addition to the potential limitations of its impact.
| Case Report | ▴Top |
This patient was a female infant born to non-consanguineous parents at 39 + 5/7 weeks (3,385 g, 51 cm). During early infancy, she exhibited signs of congestive heart failure (CHF). Transthoracic echocardiogram at 3 months of age was significant for cardiomegaly with signs of pulmonary venous congestion and both membranous and muscular ventricular septal defects (VSDs). One month later, she underwent VSD repair, which resolved her cardiac symptoms. Although resolution of her CHF was confirmed on transesophageal echo, she continued to have feeding difficulty and failure to thrive requiring nutrition via nasogastric tube.
At 7 months, patient was evaluated by pediatric gastroenterology for bloody stools noted for several months. Switching to soy-based infant formula did not resolve symptoms. A flexible sigmoidoscopy at 8 months of age revealed > 6 polyps, some about 1 cm in size supporting a diagnosis of JPS. Subsequent full colonoscopy with snare polypectomy, conducted through conventional endoscopic technique (Table 1, Fig. 1) visualized and retrieved numerous colonic polyps with histology consistent with juvenile polyps, making the diagnosis of JPS.
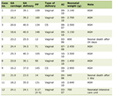 Click to view | Table 1. Endoscopic Procedures Before and After Sirolimus (Number of Polyps per Procedure by Location and Overall) |
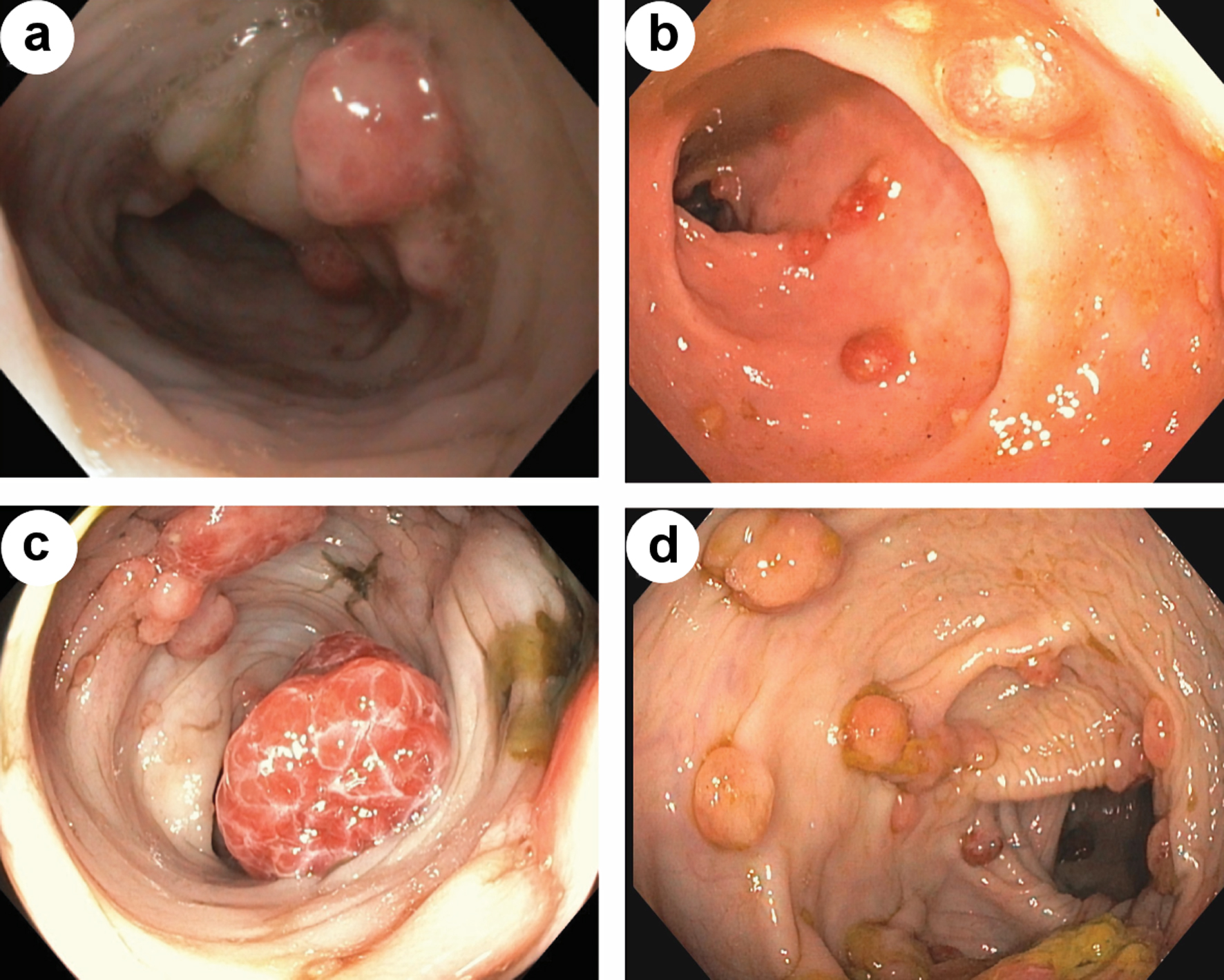 Click for large image | Figure 1. Endoscopic view of colonic polyps, prior to sirolimus initiation (a, c). Endoscopic view of colonic polyps, after start of sirolimus (b, d). |
A 17-gene hereditary colon cancer panel was sent that identified heterozygous deletions of BMPR1A and PTEN. Subsequent chromosomal microarray testing identified a 2.4 Mb deletion of 10q23.2q23.31 (arr[HG19]10q23.2q23.31(88637591_91041988)×1) containing both genes (Fig. 2). The patient had other findings consistent with PTEN hamartoma tumor syndrome, including macrocephaly, developmental delay, hypotonia, and cutaneous hemangiomas, in addition to her GI polyposis [1, 2].
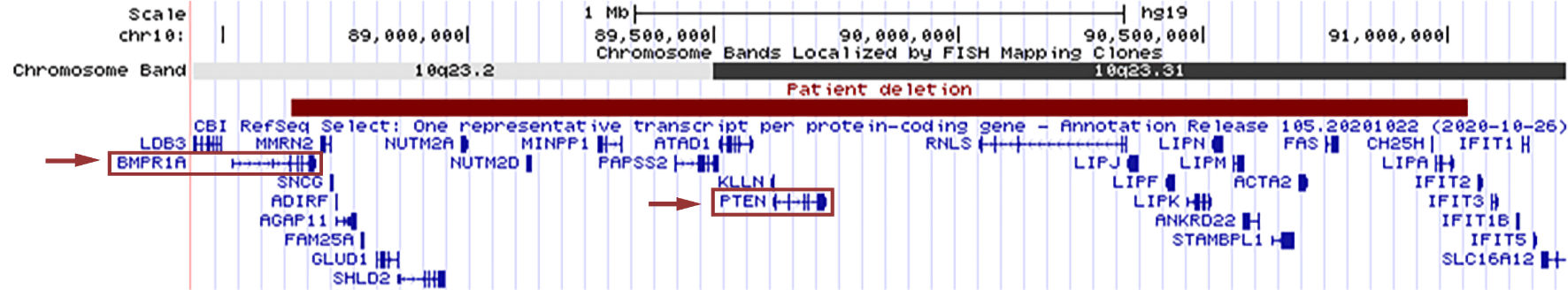 Click for large image | Figure 2. The patient’s 2.4 Mb deletion of 10q23.2q23.31 is shown in red, as represented by the UCSC Genome Browser (http://genome.ucsc.edu/index.html). RefSeq genes in the region are displayed in blue, with BMPR1A and PTEN outlined by red boxes. Red arrows indicate the gene deletions which contribute to our patient’s gastrointestinal polyposis syndrome. |
The patient had chronic hypoalbuminemia with recurrent peripheral edema and anasarca. Her clinical course included a hospitalization for severe hypoalbuminemia requiring paracentesis, albumin infusions, and blood transfusions. Workup of hypoproteinemia included elevated fecal alpha-1 antitrypsin via enzyme-linked immunoassay (ELISA) testing (> 1.13 mg/g) and reassuring synthetic liver function (prothrombin time (PT) 11 s, international normalized ratio (INR) 1.1, platelet count 162,000/µL), which were suggestive of PLE. She received intravenous immunoglobulin (IVIG) replacement for hypogammaglobulinemia, which was attributed to PLE and a potential contiguous gene deletion syndrome involving the Fas gene which can be linked to the primary immune deficiency of autoimmune lymphoproliferative syndrome (ALPS). She also required PN support via central line due to hypoalbuminemia and poor growth. She underwent an intraoperative enteroscopy with polypectomy to remove small bowel polyps contributing to intermittent intussusception.
Given the aggressive disease course, oral sirolimus therapy was started at 14 months of age with the goal of slowing the rate of polyp proliferation, limiting PLE, improving quality of life and avoiding bowel resection. Detailed information was provided to the family, including potential risks and benefits, and informed consent was obtained. Some short-term and long-term potential risks associated with sirolimus therapy include immunosuppression, dyslipidemia, pulmonary toxicity, nephrotoxicity, and impaired wound healing [8-10]. Even still, it was determined that the desired benefits of improved weight gain, reduced dependence on PN, reduced frequency of albumin and red blood cell transfusions, and avoidance of GI surgery, outweighed the risks of initiating sirolimus therapy [6, 11].
Sirolimus was started at 1.5 mg/m2 with dose adjustment targeting a serum trough level of 6 - 8 ng/mL as described previously [11]. Given the immunosuppressive properties of sirolimus, she was closely monitored for cytopenia and was maintained at an IgG trough level of 150 - 200 mg/dL with regular IgG infusions. Additional laboratory monitoring included comprehensive metabolic panel, complete blood count, and sirolimus levels every 2 weeks. Frequency of lab monitoring diminished overtime. She was also followed in the pediatric gastroenterology clinic monthly to closely monitor her clinical course.
The patient’s subsequent course included five hospitalizations for central-line associated blood stream infections, during which sirolimus was temporarily held. Her weight improved and she no longer required PN after < 1 year of starting sirolimus. Once the central line was removed, she did not require additional hospitalizations for infection.
In the 12 months following initiation of sirolimus, serum hemoglobin and albumin levels improved significantly without further need for intravenous blood products, or albumin infusions (Fig. 3). Upper endoscopy and colonoscopy with polypectomy were spaced to approximately every 6 months or longer (Table 2). Histology of polyps after initiation of sirolimus treatment remained benign and unchanged from that of the polyps before starting treatment. Despite improved albumin levels and reduced number of hospital admissions for bacteremia, trough IgG levels remained below desired levels, and she continued to require IVIG subcutaneous dosing. She continued sirolimus along with endoscopic surveillance. After > 4 years of receiving sirolimus, she remained well, taking all enteral nutrition orally and maintaining appropriate growth without the need for bowel resection.
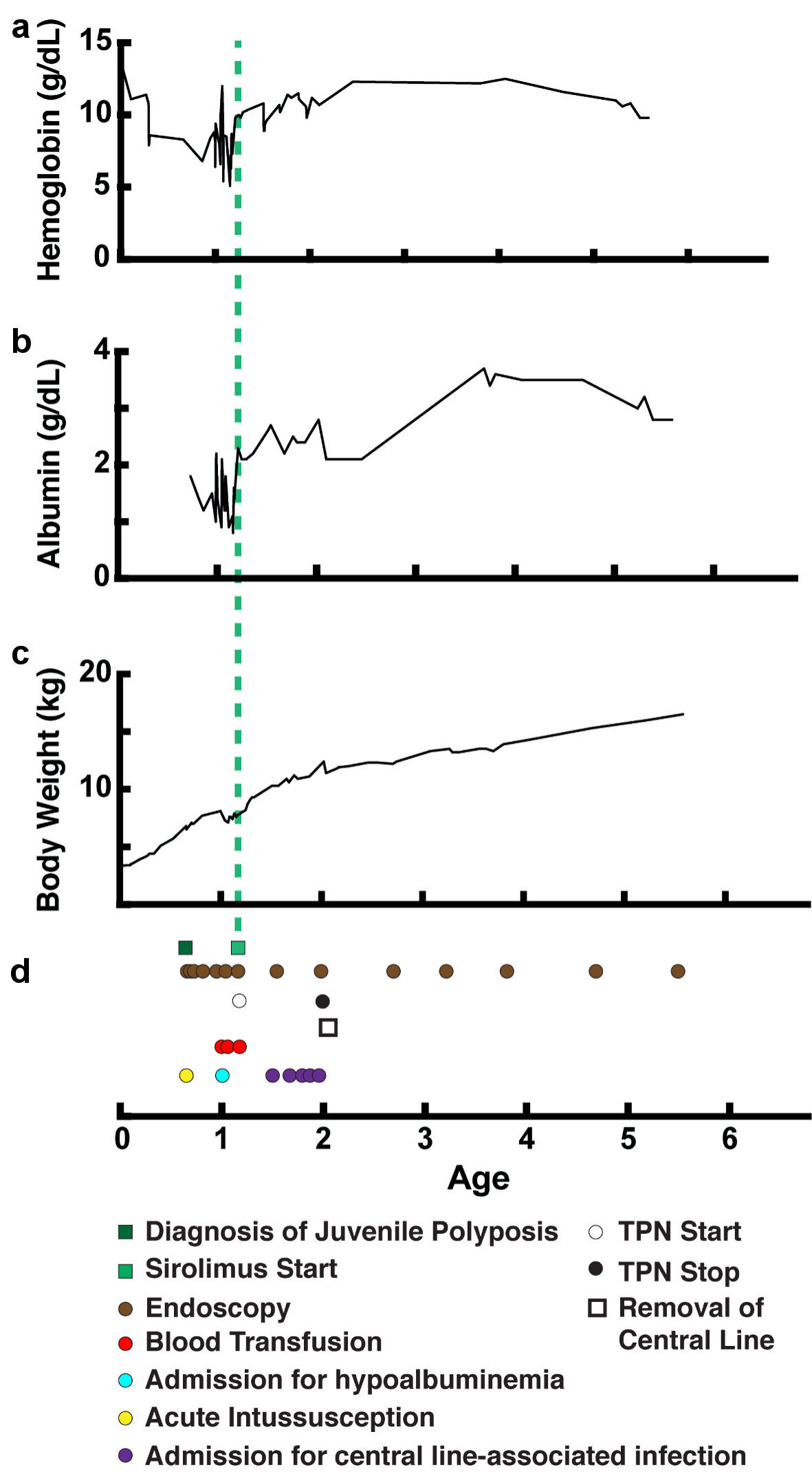 Click for large image | Figure 3. Improvement in weight gain and lab values over time with treatment. (a) Serum hemoglobin level (g/dL). (b) Serum albumin level (g/dL). (c) Body weight (kg). (d) Timeline of events (age in years). TPN: total parenteral nutrition. |
Click to view | Table 2. Reduction in Number of Polyps Resected and Frequency of Endoscopic Procedures per Year Following Initiation of Sirolimus |
| Discussion | ▴Top |
JPS is a rare autosomal dominant disease that is associated with hamartomatous growths that arise throughout the GI tract of affected children. It is defined as having ≥ 5 GI polyps, or any polyps present in a child with a family history of JPS. This syndrome is associated with increased risk of colorectal and gastric cancers in the long term but can also have a more immediate impact on health and development [1, 2]. Most patients have either a SMAD4 or BMPR1A loss of function variant. Case studies have also identified a correlation between chromosome 10q23 deletions including BMPR1A and PTEN with rare onset of a spectrum of phenotypically heterogenous polyposis syndromes [1-4].
The more aggressive form, infantile JPS, arises during early infancy, and is often associated with worse prognosis due to GI blood loss and PLE leading to comorbid sequelae such as malnutrition, severe anemia, and immune deficiency. Presence of polyps also increases risk of intussusception and GI obstruction, which may lead to emergent surgical intervention [2, 4, 6]. Compared to its less severe counterpart of JPS, infantile JPS occurs much earlier in life with more severe disease manifestations. A study of 25 JPS patients noted that those presenting in infancy had higher polyp burden, greater risk of PLE and higher need for colectomy compared to those presenting after 1 year of age (relative risk (RR) for colectomy = 2.16, 95% confidence interval (CI) = 1.02 - 5.34, P = 0.03) [5].
Sirolimus, an inhibitor of the mammalian target of rapamycin (mTOR), appears to participate in a cell proliferative pathway that is more activated in patients with PTEN mutations. This pathway contributes to cell proliferation, protein synthesis, and overall growth. Evidence suggests that mTOR activation plays a role in tumor growth in conditions similar to infantile JPS, such as Peutz-Jeghers (PJ) and Cowden syndrome. More specifically, it is believed that loss of PTEN, a tumor suppressor gene, causes dysregulation of the mTOR pathway, leading to tumorigenesis [12]. Sirolimus has reported roles in post-transplant immunosuppression, cancer treatment, and reduction of GI polyp burden in PJ syndrome in animal models [12, 13].
Prior case studies cite some success with surgical treatments with bowel resection and multi-organ transplantation, colectomy with endoscopic polypectomy, and with sirolimus with subtotal colectomy [4, 6, 7]. In a report of one JPS patient, near-disappearance of polypoid lesions was noted with sirolimus therapy, along with cessation of intravenous iron infusions, with minimal anticipated side effects of sirolimus after 1 year of treatment. In that report, the patient had a solitary mutation and age at onset of symptoms was 6 years (compared to polygenic deletions and younger age of onset in the infantile JPS patient described in this paper) [11]. In another case report, treatment of JPS with colectomy alone did not successfully mitigate PLE with ongoing need for albumin and red blood cell infusions. Subsequent treatment with sirolimus allowed the patient to discontinue recurrent transfusions [6]. This patient had polygenic mutations, like our patient [6]. While the degree of benefit appears to vary among patients, the previously reported positive outcomes, along with our findings in this case report, demonstrate a consistent observation that sirolimus can play a substantial role in the treatment of GI polypoid disease, including infantile JPS.
In this paper, we demonstrate a case of successful treatment with early use of sirolimus and aggressive endoscopic surveillance with polypectomy. This allowed for reduction in GI blood and protein loss, optimization of weight gain with successful weaning off PN while avoiding bowel resection. This is consistent with recently published data on patients with infantile JPS [5]. Even still, until this point, a full narrative account of a patient with infantile JPS, treated with sirolimus without need for bowel resection, had not been published.
With use of sirolimus, we achieved our goal of reducing GI polyp burden (as noted by reduced number of polyps resected per year, Table 2) and addressing poor weight gain, PLE and anemia, without the need for bowel resection. An additional benefit observed following sirolimus therapy was the reduced frequency of endoscopic intervention needed with polypectomy (Table 2). Given that mTOR activation has a potential role in the subsequent development of malignancy in colonic polyposis patients, we also hope that continued sirolimus use may potentially prevent malignant conversion of polyps. The histology of the patient’s polyps has remained benign [12].
We share this case as an example of successful clinical outcomes with medical and endoscopic management to provide a precedent for treatment of other patients with similar disease processes. We acknowledge that treatment with sirolimus did not fully eliminate polyp growth or the need for polypectomy; however, we believe that the improvement in weight gain, along with decreased frequency of contact with healthcare services allowed the patient to resume a life closer to normalcy in her childhood.
Learning point
Sirolimus may improve outcomes in patients with polygenic infantile JPS; however, it may not completely suppress polyp growth.
Acknowledgments
None to declare.
Financial Disclosure
None to declare.
Conflict of Interest
Authors declare no conflict of interest in the creation of this case report.
Informed Consent
Written consent was obtained from patient’s family.
Author Contributions
Lisa Bell contributed to chart review, literature review, and drafting of manuscript. John Bernat provided in-depth description of patient’s genetic workup and mutations, and reviewed draft for intellectual content. Riad Rahhal identified case of interest, did literature review, and revision of draft for intellectual content.
Data Availability
The authors declare that the data supporting the findings of this study are presented within the content of this article.
| References | ▴Top |
- Jelsig AM, Qvist N, Brusgaard K, Nielsen CB, Hansen TP, Ousager LB. Hamartomatous polyposis syndromes: a review. Orphanet J Rare Dis. 2014;9:101.
doi pubmed - Brosens LA, Langeveld D, van Hattem WA, Giardiello FM, Offerhaus GJ. Juvenile polyposis syndrome. World J Gastroenterol. 2011;17(44):4839-4844.
doi pubmed - Septer S, Zhang L, Lawson CE, Cocjin J, Attard T, Ardinger HH. Aggressive juvenile polyposis in children with chromosome 10q23 deletion. World J Gastroenterol. 2013;19(14):2286-2292.
doi pubmed - Oliveira PH, Cunha C, Almeida S, Ferreira R, Maia S, Saraiva JM, Lopes MF. Juvenile polyposis of infancy in a child with deletion of BMPR1A and PTEN genes: surgical approach. J Pediatr Surg. 2013;48(1):e33-37.
doi pubmed - Taylor H, Yerlioglu D, Phen C, Ballauff A, Nedelkopoulou N, Spier I, Loverdos I, et al. mTOR inhibitors reduce enteropathy, intestinal bleeding and colectomy rate in patients with juvenile polyposis of infancy with PTEN-BMPR1A deletion. Hum Mol Genet. 2021;30(14):1273-1282.
doi pubmed - Busoni VB, Orsi M, Lobos PA, D'Agostino D, Wagener M, De la Iglesia P, Fox VL. Successful Treatment of Juvenile Polyposis of Infancy With Sirolimus. Pediatrics. 2019;144(2):e20182922.
doi pubmed - Chandan OC, Reyes-Santiago E, Vargas L, Mercer D, Grant W, Langnas A, Quiros-Tejeira RE, et al. Multiorgan Transplantation as a Viable Treatment Option in Infantile Juvenile Polyposis Syndrome. J Pediatr Gastroenterol Nutr. 2019;68(5):e86-e87.
doi pubmed - Kuypers DR. Benefit-risk assessment of sirolimus in renal transplantation. Drug Saf. 2005;28(2):153-181.
doi pubmed - Ji L, Xie W, Zhang Z. Efficacy and safety of sirolimus in patients with systemic lupus erythematosus: A systematic review and meta-analysis. Semin Arthritis Rheum. 2020;50(5):1073-1080.
doi pubmed - Buhaescu I, Izzedine H, Covic A. Sirolimus—challenging current perspectives. Ther Drug Monit. 2006;28(5):577-584.
doi pubmed - Martin-Masot R, Cardelo Autero N, Ortiz Perez P, Torcuato Rubio E, Vazquez Pedreno L, Gallego Fernandez C, Blasco-Alonso J, et al. Sirolimus for the Treatment of Juvenile Polyposis in Childhood. ACG Case Rep J. 2021;8(8):e00646.
doi pubmed - Benjamin D, Colombi M, Moroni C, Hall MN. Rapamycin passes the torch: a new generation of mTOR inhibitors. Nat Rev Drug Discov. 2011;10(11):868-880.
doi pubmed - Wei C, Amos CI, Zhang N, Zhu J, Wang X, Frazier ML. Chemopreventive efficacy of rapamycin on Peutz-Jeghers syndrome in a mouse model. Cancer Lett. 2009;277(2):149-154.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Gastroenterology Research is published by Elmer Press Inc.