

| Gastroenterology Research, ISSN 1918-2805 print, 1918-2813 online, Open Access |
| Article copyright, the authors; Journal compilation copyright, Gastroenterol Res and Elmer Press Inc |
| Journal website http://www.gastrores.org |
Original Article
Volume 11, Number 6, December 2018, pages 404-410
Autophagy Related-Protein 16-1 Up-Regulated in Hepatitis B Virus-Related Hepatocellular Carcinoma and Impaired Apoptosis
Jiaranai Peantuma, Areerat Kunanopparatb, Nattiya Hirankarnb, Pisit Tangkijvanichc, Ingorn Kimkonga, d, e
aDepartment of Microbiology, Faculty of Science, Kasetsart University, Bangkok, Thailand
bCenter of Excellence in Immunology and Immune Mediated Diseases, Department of Microbiology, Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand
cResearch Unit of Hepatitis and Liver Cancer, Department of Biochemistry, Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand
dCenter for Advanced Studies in Tropical Natural Resources, National Research University-Kasetsart University, Bangkok, Thailand
eCorresponding Author: Ingorn Kimkong, Department of Microbiology, Kasetsart University, 50 Ngam Wong Wan Rd., Ladyaow Chatuchak, Bangkok 10900, Thailand
Manuscript submitted August 7, 2018, accepted October 2, 2018
Short title: ATG16L1 and Hepatocellular Carcinoma
doi: https://doi.org/10.14740/gr1075w
| Abstract | ▴Top |
Background: Hepatocellular carcinoma (HCC) as primary malignancy of the liver has become the most common type of cancer worldwide. HCC development is mainly caused by viruses, especially the hepatitis B virus (HBV). Autophagy is an important defense mechanism against virus infection; however, HBV promotes autophagy mediated by the HBx protein which stimulates its replication. The autophagy-related protein 16-1 (ATG16L1) binds to the ATG12-ATG5 conjugate and forms a large protein autophagosome complex. Previous studies indicated that the ATG12-ATG5 conjugate was involved in HBV-associated HCC. Therefore, the ATG16L1 protein might consistently relate to this condition.
Methods: Accordingly, the ATG16L1 protein expression was determined in tumor and non-tumor liver cell lines and liver tissue samples using immunoblotting, and also investigated in ATG16L1-knockdown cells to further clarify this function.
Results: Our results showed that the ATG16L1 protein was up-regulated in HepG2.2.15 and HepG2 cell lines compared to THLE-2 cells. This protein also increased in tumor liver tissues of HCC patients with HBV infection compared to adjacent non-tumor tissues. Silenced-ATG16L1 also significantly promoted apoptosis in HepG2 cells cultured in starvation conditions.
Conclusions: Findings suggested ATG16L1 as an important molecule involved in apoptosis processes for HCC cells. A more profound understanding is required regarding the mechanisms that link autophagy and apoptosis in HCC development.
Keywords: Autophagy-related protein 16-1; Autophagy; Hepatitis B virus; Hepatocellular carcinoma
| Introduction | ▴Top |
Hepatocellular carcinoma (HCC) is a primary malignancy of the liver that can be caused by various factors including chronic alcohol consumption, non-alcoholic fatty liver disease, obesity, toxic exposure, hepatitis C virus and most especially the hepatitis B virus (HBV) [1]. HBV infection is a viral infection that can be spread through blood and other bodily fluids and attacks the liver to induce acute and chronic diseases. HBV is widespread in spite of the availability of an effective vaccine and antiviral treatments. The World Health Organization (WHO) estimated that approximately 887,000 people die worldwide annually due to HBV infection, while some 257 million are diagnosed with remaining detectable HBsAg particles in serum and liver tissue. In addition, a previous epidemiology study reported that an estimated 240 million people are chronically infected with this virus [2]. Autophagy or self-eating is a catabolic process characterized by the lysosomal degradation of materials and/or organelles to maintain cellular functions such as cellular homeostasis, differentiation, development, and survival. Therefore, stress or starvation condition can result in degradation processes to recycle energy [3]. More importantly, circumstances of autophagy deficiency lead to the pathogenesis of diverse disease states as myopathy, neuronal degeneration, microbial infection, inflammatory bowel disease, aging, and especially cancer [4].
The autophagy-related protein 16-1(ATG16L1) is a member of the autophagy-related protein family and plays a critical role in the autophagy pathway via interaction with the ATG12-ATG5 molecular conjugate complex to produce a bilayer membrane for elongation and autophagosome formation. These three molecules may function to consistently produce the autophagy process. Interestingly, a previous study revealed that increasing the ATG12-ATG5 conjugate in HCC was associated with HBV replication both in cell lines and liver tissues, resulting in a noticeable correlation to the apoptosis pathway [5]. Moreover, expression of the ATG16L1 gene level significantly increased in HepG2.2.15 and HepG2 cells compared to primary normal liver cells (THLE-2) [6]. To support this evidence we examined ATG16L1 protein levels in THLE-2, HepG2, and HepG2.2.15 cells, as well as in non-HBV and HBV-infected HCC tissues. We also analyzed the functional role of ATG16L1 in hepatocarcinogenesis.
| Materials and Methods | ▴Top |
Study design
To investigate the role of ATG16L1 in HCC, the expression of this molecule is examined in two HCC cell lines in relative to one normal liver cell line. HepG2 (human liver cancer cell HepG2; ATCC® HB8065TM, Singapore) and HepG2.2.15 (derived from the complete HBV genome stable transfected HepG2) were obtained from Professor Antonio Bertoletti (Singapore Institute for Clinical Sciences, A*Star) to determine tumorigenic results. THLE-2 (normal liver epithelial cell; ATCC® CRL-2706™, Manassas, VA, USA) was cultured with complete growth Bronchial Epithelial Cell Growth Medium, BulletKit™ (BEGM, Lonza) and both human liver cancer cells were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco) supplemented with 10% fetal bovine serum (FBS) and antibiotics. To maintain the HBV plasmid in HepG2.2.15, 150 µg/mL of Geneticin® (G418 Sulfate; Gibco) was added into the culture media. These three cells were induced to starvation condition by handling in Earle’s balanced salt solution (EBSS; Invitrogen) for 4 h in a humidified incubator at 37 °C with 5% CO2.
Fifteen paired HCC tissue samples, including tumor liver tissues and their corresponding adjacent non-tumor liver tissues, were divided into two groups as non-HBV (N = 7) and HBV-infected (N = 8) liver tissues. Samples were collected from patients at King Chulalongkorn Memorial Hospital and the procedure was approved by the Institutional Review Board (IRB No. 396/55). All HCC tissue samples were diagnosed by histology, snap-frozen in liquid nitrogen and then conserved at -80 °C until required for western blot analysis.
Study assessments
Cell and HCC liver tissue samples were lysed in radioimmunoprecipitation assay buffer (RIPA buffer) added with protease inhibitor and applied by sonicator. Protein concentration was subsequently quantified using Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA, USA) compared to the protein standard. Whole protein was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred onto polyvinylidene difluoride (PVDF) membrane. All experiments were probed with monoclonal antibodies: ATG16L1, GAPDH and applied to the secondary antibody conjugated with horseradish peroxidase-linked anti-rabbit. The target protein was visualized by enhanced typical chemiluminescence (ECL) and resulted in band intensity.
The cell line was transfected with the set of shRNA ATG16L1 expression vectors using Lipofectamine® 2000 Transfection Reagent (Invitrogen) diluted with Opti-MEM® I Reduced Serum Medium (Invitrogen). The ratio for cell number and transfected reagent in our experiments was 2×105 seeding cells per 2.5 µL of 1 mg/mL Lipofectamine® dissolved in 100 µL Opti-MEM®, incubated at 25 °C for 30 min. The transfection mixture was gently dropped into cell culture plates and green fluorescence protein was monitored under an inverted fluorescence microscope after 24 h to observe the efficiency of transfection.
RNA was extracted using Real Genomics Total RNA Extraction Kit (RBC Bioscience) and 1 µg RNA concentration was converted to cDNA using a High-Capacity Reverse Transcription Kit (Applied Biosystems) following the manufacturer’s guidelines. An ABI Thermal Cycler 7500 Real-Time PCR Instrument (Applied Biosystems) was used to analyze ATG16L1 mRNA expression patterns by a commercial (2X) Power SYBR® Green PCR Master Mix (Applied Biosystems). Two primer pairs were obtained from the study of Tantithavorn V et al [6] as ATG16L1; forward 5’-AGGGTTCCCTATCTGGCAGT-3’, reverse 5’-GATTCGGCTTGCAAAATCAT-3’ and beta-actin; forward 5’-ACCAACTGGGACGACATGGAGAA-3’, reverse 5’-GTGGTGGTGAAGCTGTAGCC-3’. The ATG16L1 mRNA expression level was calculated by the 2-ΔΔCt method.
Genomic DNA was extracted from shRNA ATG16L1 transfected HepG2.2.15 cells (HBV-stable transfected HCC cell line) under starvation condition, performed by a commercial (2X) Power SYBR® Green PCR Master Mix (Applied Biosystems) using an ABI Thermal Cycler 7500 Real-Time PCR Instrument, and compared to the standard PreS1 copy number. To analyze HBV DNA level, a pair of specific primers was used as forward 5’-GGGTCACCATATTCTTGGGAAC-3’ and reverse 5’-CCTGAGCCTGAGGGCTCCAC-3’ [5].
Cell viability in proliferation was measured by CellTiter96® AQueous One Solution Reagent. HepG2 and HepG2.2.15 were transiently transfected with shRNA ATG16L1 plasmids for 96 h in triplicate independent experiments using DMEM as a colorimeter calibration. Twenty µL of MTS solution was directly added into 100 µL cell culture medium and the plate was maintained at 37 °C with 5% CO2 for 3 h before measuring the quantity of formazan. This MTS soluble product was determined at 48, 72 and 96 h at the absorbance spectrum of 490 nm following the product technical instructions.
A multicolor flow cytometer was used to assess apoptosis in HepG2 and HepG2.2.15 cell populations. The apoptosis process is characterized by certain morphological features. After washing the cells with cold PBS twice, we first fixed two kinds of this cell with BD™ Annexin V/APC and eFluor780/APC-cy7 to distinguish between apoptotic and death cells. This independent experiment was performed by staining cells for 30 min on ice in the dark room and then diluting with an optimal volume of cold PBS for flow cytometry analysis.
Statistical analysis
Optical density of the protein signals was calculated using the Image Studio Lite program and significant differences were determined with GraphPad Prism, version 5.0 software (San Diego, CA, USA). A threshold of P < 0.05 was defined as statistically significant.
| Results | ▴Top |
Expression profile of ATG16L1 protein in HCC cell lines and tissues
The protein expression of ATG16L1 in HepG2.2.15, HepG2 and THLE-2 was investigated using Western blotting. Results revealed that this protein increased in HepG2.2.15 and HepG2 cells compared to the THLE-2 cell line (Fig. 1a). For the HCC tissue samples, six out of seven non-HBV HCC tissue samples showed down-regulation, and seven out of eight HBV-infected HCC tissues exhibited up-regulation of ATG16L1 protein (Fig. 1b). ATG16L1 protein was significantly up-regulated in the HBV-infected HCC tissue group (P = 0.0410), but not in non-HBV infected HCC tissues, (P = 0.0739) (Fig. 1c). Hence under this circumstance, ATG16L1 may play a role in HBV-associated HCC.
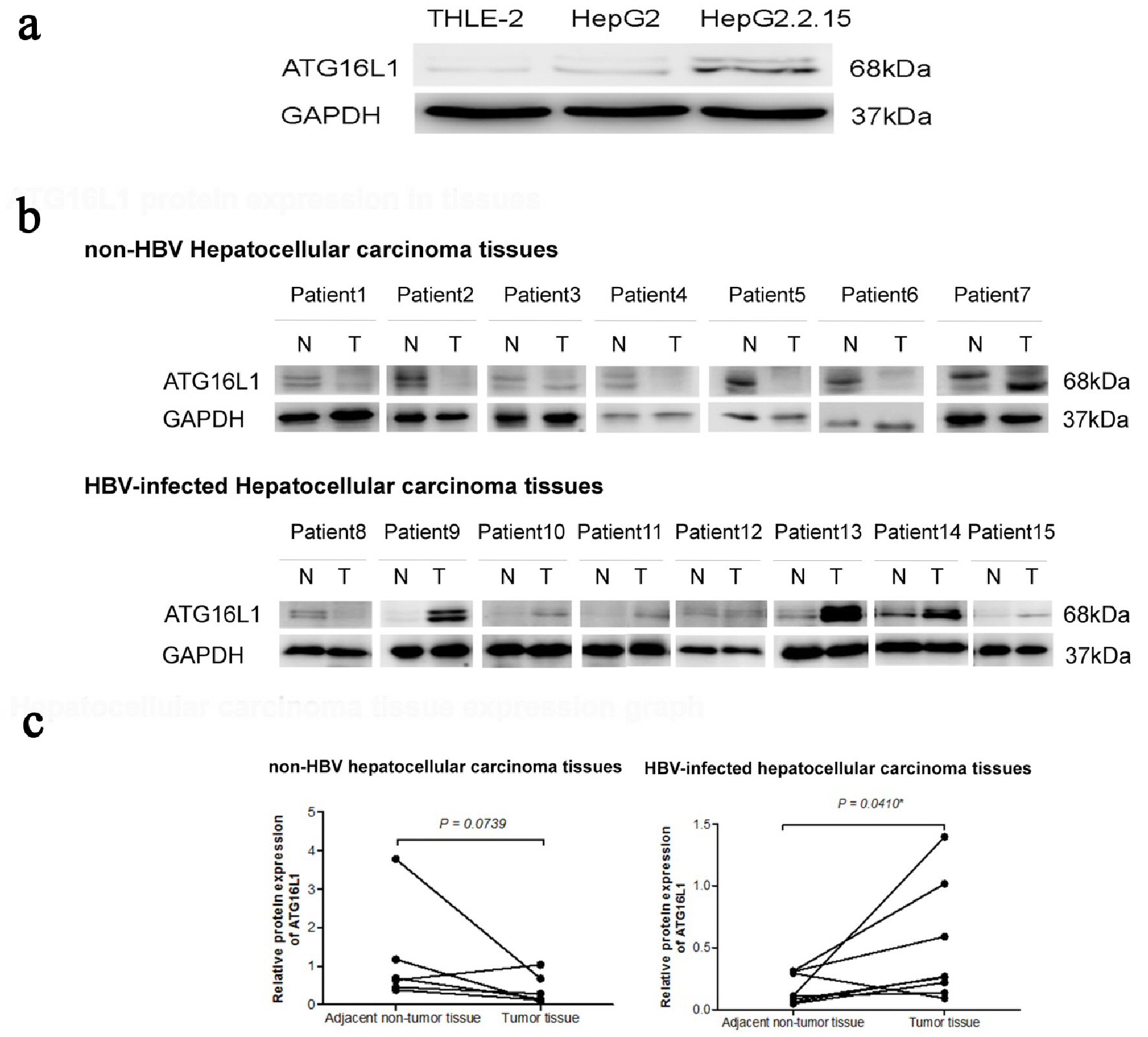 Click for large image | Figure 1. ATG16L1 becomes visible in the perpetual double bands at approximately 68 kDa as alpha (upper) and beta (lower) forms. (a) ATG16L1 protein expression in THLE-2, HepG2 and HepG2.2.15 cell lines, respectively. (b) Protein level in HCC liver tissues (denoted as T) and their corresponding adjacent non-tumor tissues (denoted as N). (c) The quantitative result of HCC liver tissues showing the statistic value. All results were achieved by immunoblotting analysis using monoclonal ATG16L1 antibody (68 kDa) with GAPDH used as loading control (37 kDa) under starvation condition. |
ATG16L1 gene and protein knockdown efficiency
We first verified the knockdown efficiency by observing green fluorescent protein (GFP) expression at 72 h post-transfection. Obviously, we found GFP expression exhibited under fluorescence microscope both in HepG2 and HepG2.2.15 cells. Our knockdown gene results indicated that ATG16L1 was successfully silenced when transfected with shRNA ATG16L1 clone 3 plasmid (Fig. 2a). To guarantee the efficiency of shATG16L1 knocked down, Western blot analysis was also used to detect ATG16L1 expression in HepG2 and HepG2.2.15 cells (Fig. 2b).
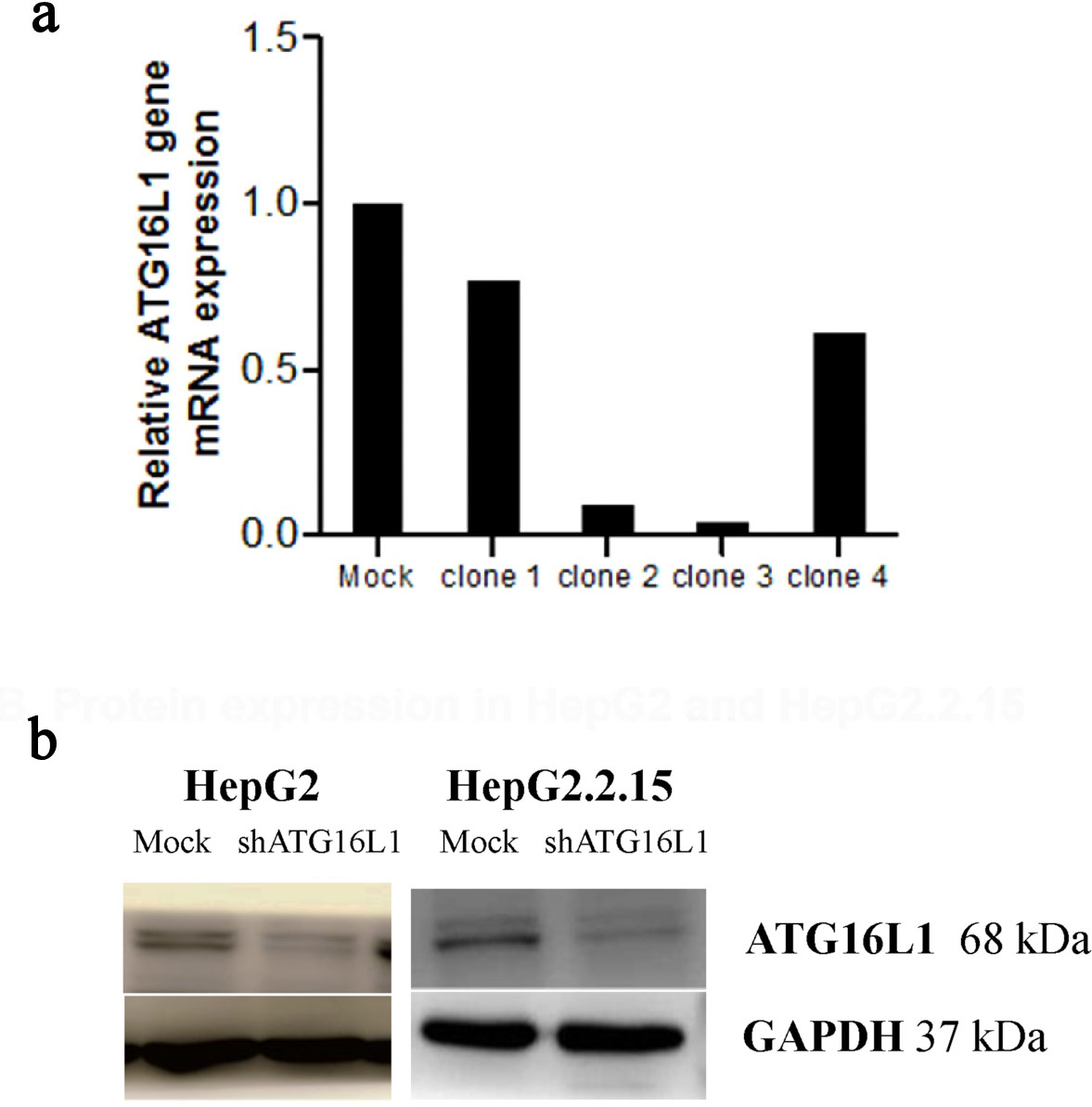 Click for large image | Figure 2. Plasmid clone collection. (a) ATG16L1 mRNA level in shRNA ATG16L1 clone 1-4 with its mock. (b) Decreased protein of shATG16L1 clone 3 in HepG2 and HepG2.2.15. |
ATG16L1 and hepatitis B virus replication
Our protein results showed that ATG16L1 increased in HBV-HCC cells. Therefore, we determined the effect of this protein on HBV survival by quantifying HBV genomic DNA in the silenced cells. Our results showed no significant statistical difference between HBV copy number titer in shATG16L1-transfected HepG2.2.15 cells compared to the mock control (Fig. 3). Thus, ATG16L1 may not be involved in HBV replication in HCC cells.
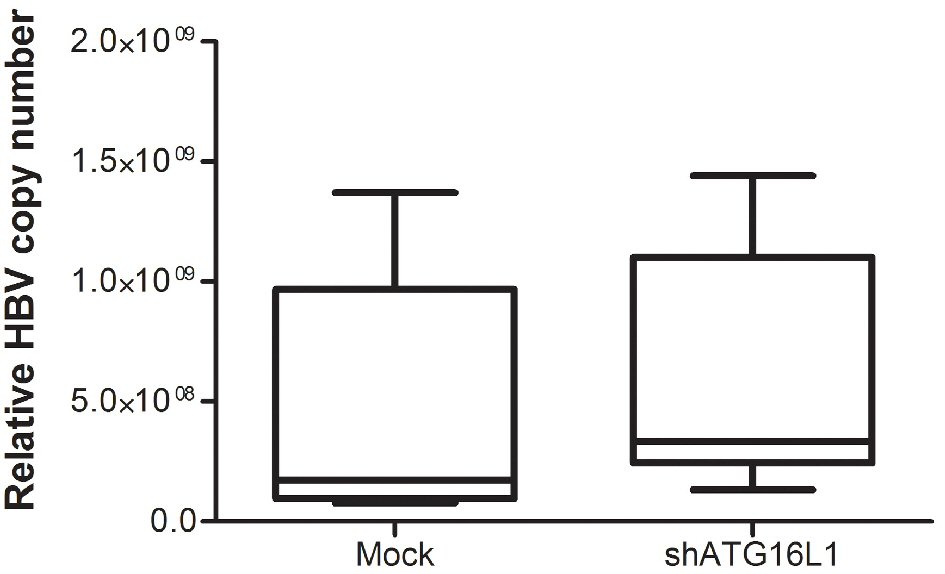 Click for large image | Figure 3. After starvation, HBV titer was amplified using SYBR green signal detection against preS1 specific primers in the HepG2.2.15 cell line. The assay was assessed using HBV copy number standard compared to shATG16L1 knockdown cell and its mock control. All results were performed as triplicate independent experiments. |
ATG16L1 and cell proliferation
In HCC, cell proliferation is regulated by the cell cycle which cannot control cell growth or cell division and results in unlimited proliferation. Therefore, we examined whether ATG16L1 knocked-down might affect the ability of cell proliferation. Our findings showed no significant differences for percentage cell proliferation in shATG16L1-transfected HepG2 (Fig. 4a) and shATG16L1-transfected HepG2.2.15 cells compared with the mock control (Fig. 4b). Thus, ATG16L1 may not play an important role in cell proliferation for both HCC and/or HBV-infected HCC cells.
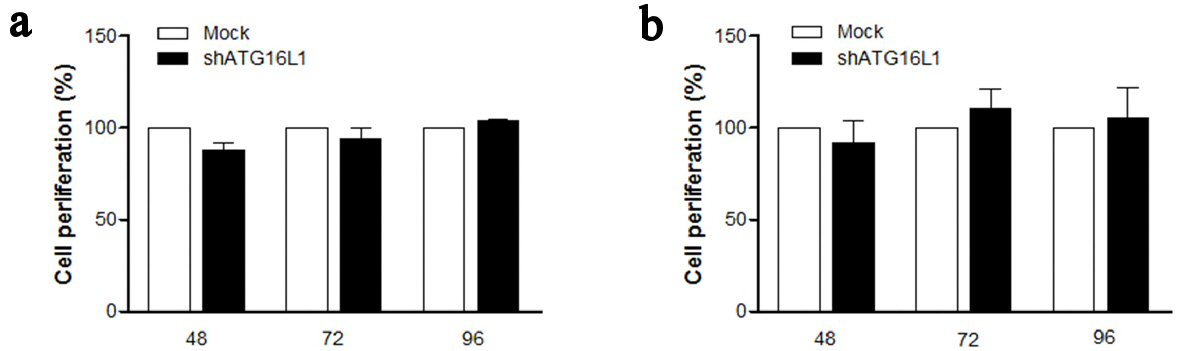 Click for large image | Figure 4. ATG16L1 knock down does not affect cell proliferation in HepG2 (a) and HepG2.2.15 (b) at 48, 72, and 96 h. MTS assay was used as the colorimetric method (490 nm) to determine the number of cell progressions after transfecting shATG16L1 plasmid for 24 h. Absorbance was calculated as cell proliferation percentage and shown as the relative ratio with its mock. All results were performed as triplicate independent experiments. |
Role of ATG16L1 in apoptosis
To better understand the self-destruction control in liver cancer cells, we considered apoptosis in cells with silenced ATG16L1 (Fig. 5a). Interestingly, our results demonstrated that apoptosis percentage significantly increased in HepG2 cells transfected with shATG16L1 plasmids at 72 h post-transfection in starvation condition (P = 0.0173). Conversely, HepG2.2.15 cells showed decreasing apoptosis but this was not statistically significant (Fig. 5b). This result suggested that HBV in HepG2.2.15 cells might not be able to utilize ATG16L1 for controlling cell growth or apoptosis. Overall, our data indicated that ATG16L1 may be an essential molecule for tumor survival in HCC without HBV.
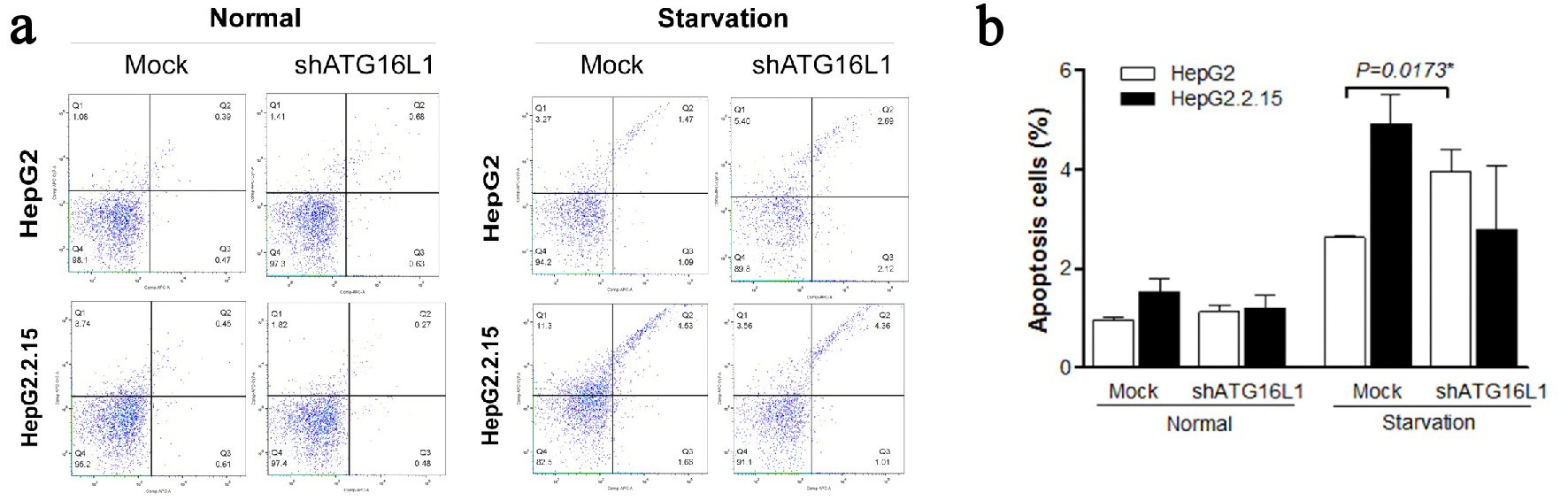 Click for large image | Figure 5. (a, b) ATG16L1 knock down in HepG2 and HepG2.2.15 leads to increase apoptosis in HepG2 cells under starvation, but not in HepG2.2.15 cells under starvation. |
| Discussion | ▴Top |
The important role of autophagy in liver cancer can be defined as tumor survival under severe deficiency in malnutrition. Additionally, it indicates dual functions in HCC as a tumor suppressor by inhibiting inflammation, P62 accumulation, oxidative stress response and genomic instability in the initiation stage and interrupting autophagic cell death in HCC promotion and progression which is called tumorigenesis [7]. In addition, previous studies suggested the HCC-HBV implication that HBV enhances and uses the autophagy process for replication via the HBx protein, leading to HCC [8]. Results from autophagy protein research have defined ATG16L1 as a necessary protein for the ATG12-ATG5 conjugate complex to mediate phosphatidylethanolamine (PE) which is important for controlling autophagosome elongation [9]. Interestingly, ATG12 and ATG5 are involved in HBV-associated HCC [5].
In this study, we were interested in ATG16L1 protein and our results suggested that ATG16L1 might be involved in HCC and/or HBV due to high levels of ATG16L1 protein in both HBV-infected HCC cell lines and tissues compared to the control cells/tissues. These protein expression profiles were consistent with results from a previous ATG16L1 mRNA study that demonstrated ATG16L1 was up-regulated in HepG2.2.15 and HepG2 cells compared with the THLE-2 cell line, although the mRNA level did not differ in liver tissues [6]. With this fact established beyond doubt, we proceeded to observe HBV replication in cells with silenced ATG16L1 by verifying viral genomic DNA in HepG2.2.15 cells. Our findings exhibited no change of HBV titer in HepG2.2.15 cells with silenced ATG16L1 compared with its mock control. The result suggested that ATG16L1 may not assist HBV replication in HCC. Therefore, HBV may interact with other molecules to benefit its replication. Previous study showed that HBV replication was related to ATG12 via IFN pathway in HCC [10]. Although ATG16L1 was identified together with ATG12-5 as ATG12-5-16L1 complex, but the function of this autophagy molecule is different from ATG12-5. However, ATG16L1 might correlate with other functions in HCC.
In a previous report, the percentage of ATG12-ATG5 gene disruption in HepG2 and HepG2.2.15 apoptotic cells significantly increased in both cell lines. Hence, these autophagy genes link with the apoptosis pathway in HBV and/or HCC cells [5]. We initially studied formazan production in primary screening of cell proliferation. Our MTS assay results determined that the number of cell proliferations in both shATG16L1-transfected cell lines showed no statistical differences. Therefore, ATG16L1 might not be the molecule encouraging proliferation of HCC and HBV-HCC cells. However, in our apoptosis study, there was a significant apoptosis increment in HepG2 cells when we silenced with shATG16L1 under starvation condition; in turn, apoptosis rate was suppressed in HepG2.2.15 cells but not with a statistically significant value. Our findings indicated that ATG16L1 is important for the apoptosis pathway in HCC.
Interestingly, previous studies suggested that many ATG proteins (ATGs) act as linkers between apoptosis and autophagy. For instance, ATG3 can conjugate with ATG12 as a complex to regulate mitochondria homeostasis and cell death, while ATG4, in ATG4D form, cleaved into two fragments which can lead to autophagosome reduction and promote apoptosis [11]. ATGs might, therefore, somehow interact with other molecules in the apoptosis pathway similar to ATG12 functions and inactivate Bax; a critical effector of apoptosis and cytochrome c release [12]. ATG5 was also considered to be involved in the interplay of Bcl-XL autophagy-apoptosis crosstalk [13]. The literature review of ATG16L1 and apoptosis interaction in HCC is insufficient to support our data. One study suggested that ATG16L1 could play a critical role and interact with EVA1A or TMEM166, contributing to autophagy and apoptosis induction [14]. ATG16L1 is one of the members of the ATG12-5/16L1 complex; therefore, it might relate with the Bcl-2 family, similarly to ATG12 and ATG5. Further studies are needed to investigate the mechanism of ATG16L1-mediated inhibition of apoptosis in HCC.
Conclusions
In conclusion, our results indicated ATG16L1 to be an essential autophagy protein, involved with the apoptosis pathway in HCC. This protein may play a key role as a mediate molecule that acts as a link between autophagy and apoptosis. However, further research is required to clarify this function of ATG16L1.
Acknowledgments
This study was supported by a research grant from Kasetsart University Research and Development Institute (KURDI), Faculty of Science and the Graduate School, Kasetsart University.
Conflict of Interest
There is no conflict of interest declaration to disclose.
| References | ▴Top |
- El-Serag HB, Rudolph KL. Hepatocellular carcinoma: epidemiology and molecular carcinogenesis. Gastroenterology. 2007;132(7):2557-2576.
doi pubmed - Ott JJ, Stevens GA, Groeger J, Wiersma ST. Global epidemiology of hepatitis B virus infection: new estimates of age-specific HBsAg seroprevalence and endemicity. Vaccine. 2012;30(12):2212-2219.
doi pubmed - Fang K, Chen SP, Lin CW, Cheng WC, Huang HT. Ellipticine-induced apoptosis depends on Akt translocation and signaling in lung epithelial cancer cells. Lung Cancer. 2009;63(2):227-234.
doi pubmed - Chen N, Karantza-Wadsworth V. Role and regulation of autophagy in cancer. Biochim Biophys Acta. 2009;1793(9):1516-1523.
doi pubmed - Kunanopparat A, Kimkong I, Palaga T, Tangkijvanich P, Sirichindakul B, Hirankarn N. Increased ATG5-ATG12 in hepatitis B virus-associated hepatocellular carcinoma and their role in apoptosis. World J Gastroenterol. 2016;22(37):8361-8374.
doi pubmed - Tantithavorn V, Hirankarn N, Tangkijvanich P, Kimkong I. Expression profile of ATG16L1 and mTOR Genes in Hepatitis B Virus Infection. Proceedings of 53rd Kasetsart University Annual Conference: Science, Genetic Engineering, Architecture and Engineering, Agro-Industry, Natural Resources and Environment. 2015;1412(53):93-100.
- Lee YJ, Jang BK. The role of autophagy in hepatocellular carcinoma. Int J Mol Sci. 2015;16(11):26629-26643.
doi pubmed - Tang SW, Ducroux A, Jeang KT, Neuveut C. Impact of cellular autophagy on viruses: Insights from hepatitis B virus and human retroviruses. J Biomed Sci. 2012;19:92.
doi pubmed - Klein KA, Jackson WT. Picornavirus subversion of the autophagy pathway. Viruses. 2011;3(9):1549-1561.
doi pubmed - Kunanopparat A, Hirankarn N, Kittigul C, Tangkijvanich P, Kimkong I. Autophagy machinery impaired interferon signalling pathways to benefit hepatitis B virus replication. Asian Pac J Allergy Immunol. 2016;34(1):77-85.
pubmed - Li M, Gao P, Zhang J. Crosstalk between Autophagy and Apoptosis: Potential and Emerging Therapeutic Targets for Cardiac Diseases. Int J Mol Sci. 2016;17(3):332.
doi pubmed - Rubinstein AD, Eisenstein M, Ber Y, Bialik S, Kimchi A. The autophagy protein Atg12 associates with antiapoptotic Bcl-2 family members to promote mitochondrial apoptosis. Mol Cell. 2011;44(5):698-709.
doi pubmed - Yousefi S, Perozzo R, Schmid I, Ziemiecki A, Schaffner T, Scapozza L, Brunner T, et al. Calpain-mediated cleavage of Atg5 switches autophagy to apoptosis. Nat Cell Biol. 2006;8(10):1124-1132.
doi pubmed - Hu J, Li G, Qu L, Li N, Liu W, Xia D, Hongdu B, et al. TMEM166/EVA1A interacts with ATG16L1 and induces autophagosome formation and cell death. Cell Death Dis. 2016;7(8):e2323.
doi pubmed
This article is distributed under the terms of the Creative Commons Attribution Non-Commercial 4.0 International License, which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Gastroenterology Research is published by Elmer Press Inc.